CYP2D6抑制剂对艾瑞昔布大鼠体内药代动力学的影响*
2020-04-11何文娟王伟美张志清
何文娟,王伟美,贡 莹,张志清△
(1.河北医科大学第二医院,河北 石家庄050000;2.哈励逊国际和平医院,河北 衡水053000)
艾瑞昔布为环氧合酶(COX)-2抑制剂,为我国自主研发的化学药品1.1类新药[1]。其通过抑制COX-2 mRNA的表达而发挥抗炎作用[2-3],用于缓解骨关节炎的疼痛症状。CHEN等[4]研究表明,艾瑞昔布具有与罗非昔布相当的抗炎作用。帕罗西汀作为一种选择性5-羟色胺(5-HT)再摄取抑制剂,广泛用于治疗抑郁症和强迫症等疾病[5-6]。帕罗西汀既是细胞色素P450酶(CYP)2D6的底物,又是其强抑制剂,但对其他P450酶很少产生影响。因此常作为CYP2D6的选择性抑制剂用于药物相互作用的研究[7-10]。目前,有关艾瑞昔布体内代谢机制的研究尚不完善。本研究中选择帕罗西汀作为CYP2D6的选择性抑制剂,比较合用帕罗西汀前后艾瑞昔布在大鼠体内药代动力学的变化,考察CYP2D6抑制剂对艾瑞昔布在大鼠体内药代动力学的影响,进一步考察艾瑞昔布是否通过CYP2D6进行代谢,从而完善艾瑞昔布体内代谢机制的研究。现报道如下。
1 仪器、试药与动物
1.1 仪器
API 4000 Plus型液相色谱-三重四级杆质谱联用仪,配有电喷雾离子化源和Analyst 1.6.1数据处理系统(AB Sciex公司);Prominence CBM-20A型液相色谱仪,配有二元梯度泵、在线脱气机、自动进样器和柱温箱(日本岛津公司)。
1.2 试药
艾瑞昔布对照品(江苏恒瑞医药股份有限公司,批号为66311003,纯度99.6%);甲苯磺丁脲对照品(大连美仑生物技术有限公司,批号为20110601,纯度99.4%);艾瑞昔布片(江苏恒瑞医药股份有限公司,批号为13052151,规格为每片0.1 g);盐酸帕罗西汀片(葛兰素史克有限公司,批号为13060410,规格为每片20 mg);甲酸和乙腈为色谱纯,乙酸乙酯为分析纯,水为屈臣氏蒸馏水。
1.3 动物
健康雄性SD大鼠40只,清洁级,体质量(220±20)g,由河北省动物实验中心提供,动物合格证号为1401040。所有实验操作均符合动物实验伦理学要求。
2 方法与结果
2.1 色谱条件
色谱柱:Diamonsil C18柱(150 mm×4.6 mm,5 μm);柱温:30℃;流动相:甲醇-水-甲酸(85∶15∶0.1,V/V/V);等度洗脱;流速:1.0 mL/min;进样量:10 μL。
2.2 质谱条件
离子化方式:电喷雾离子化源(ESI);监测方式:正离子监测;扫描方式:多反应监测(MRM);离子源喷射电压(IS):5500 V;离子源温度:550℃;气帘气(CUR)压力:15 psi;雾化气(GS1,N2)压力:40 psi;辅助气(GS2,N2)压力:40 psi;碰撞气压力:6 psi;艾瑞昔布的DP和CE值分别为100 V和45 eV,甲苯磺丁脲的DP和CE值分别为22 V和32 eV;用于定量分析的离子反应分别为m/z:370.1→236.2(艾瑞昔布)和m/z:270.1→155.2(甲苯磺丁脲)。
2.3 灌胃液制备
艾瑞昔布灌胃液:将艾瑞昔布片研成粉末,取约550mg(相当于艾瑞昔布110mg),精密称定,溶于1‰羧甲基纤维素溶液25 mL中,得质量浓度约为4.40 mg/mL的艾瑞昔布灌胃液。
帕罗西汀灌胃液:将帕罗西汀片研成粉末,取帕罗西汀粉末约50 mg(相当于帕罗西汀3.1 mg),精密称定,溶于1‰羧甲基纤维素溶液7 mL中,得质量浓度约为0.44 mg/mL的帕罗西汀灌胃液。
2.4 血浆样品处理
取血浆200 μL,置5 mL离心管中,加内标溶液20 μL,涡旋混匀,加提取剂乙酸乙酯1 mL,涡旋混匀2 min,4 500×g离心6 min,取上清液置1.5 mL离心管中,40℃水浴氮气吹干,400 μL流动相复溶,10 μL进样测定。
2.5 方法学考察
专属性试验:取艾瑞昔布对照品溶液和内标溶液分别进样,得艾瑞昔布和内标甲苯磺丁脲色谱图;分别取空白血浆、大鼠给药后1 h的血浆样品各200 μL,按2.4项下方法处理(其中空白血浆不加内标),进样,得空白血浆色谱图及血浆样品色谱图。详见图1。结果表明,内源性杂质对艾瑞昔布和内标的测定无影响。

图1 高效液相色谱质谱图
线性关系考察和最低定量限确定:制备艾瑞昔布质量浓度依次为10,20,40,80,160,200,400 ng/mL的模拟血浆样品,按2.4项下方法处理,按拟订色谱条件进样测定,记录艾瑞昔布和甲苯磺丁脲峰面积,以峰面积比值(Y)为纵坐标、艾瑞昔布血浆浓度(X)为横坐标进行线性回归,得回归方程Y=0.016X+0.072,r=0.999 7(n=7)。结果表明,艾瑞昔布质量浓度线性范围为10~400 ng/mL,最低定量限为10 ng/mL。
回收试验和精密度试验:取空白血浆15份,分为3组,分别制备低、中、高(20,80,320 ng/mL)3种质量浓度的艾瑞昔布模拟血浆样品各5份,按2.4项下方法处理并分析,记录艾瑞昔布和甲苯磺丁脲峰面积,并根据线性回归方程计算艾瑞昔布的质量浓度,该质量浓度与理论质量浓度的比值即方法回收率;于同日内处理并测定,考察日内精密度;连续5 d处理并测定,考察日间精密度。另取等量的艾瑞昔布和甲苯磺丁脲对照品溶液各20 μL,进样测定,记录峰面积;模拟血浆样品中药物峰面积与等量对照品溶液峰面积的比值,即提取回收率。结果见表1。
稳定性试验:制备质量浓度为80 ng/mL的艾瑞昔布模拟血浆样品6份,分为两组。一组即刻处理并测定,一组反复冻融3次后处理并测定,考察其反复冻融的稳定性;一组即刻处理并测定,一组于-40℃保存30 d后处理并测定,考察低温保存的稳定性;一组按2.4项下方法处理后即刻进样测定,另一组按2.4项下方法处理后于4℃放置12 h进样测定,考察处理后的放置稳定性。结果的RSD分别为2.1%,3.9%,2.1%(n=3),表明在上述条件下艾瑞昔布的稳定性良好。
表1 回收试验和精密度试验结果(±s,n=5)

表1 回收试验和精密度试验结果(±s,n=5)
名称艾瑞昔布RSD(%)日内1.81 2.21 5.90日间1.13 2.38 8.19images/BZ_18_678_1699_714_1762.png±s,%)102.2±8.07 98.71±2.76 97.01±1.84质量浓度(μg/mL)20 80 320 500方法回收率(内标提取回收率(%)61.65±1.79 65.33±3.27 72.57±2.39 51.84±1.71
2.6 药代动力学试验
将健康雄性SD大鼠40只随机分为两组,各20只,实验组连续7 d灌胃帕罗西汀灌胃液(2 mg/kg),对照组灌胃1‰羧甲基纤维素溶液,每天固定时间给药。第8天,每只大鼠灌胃艾瑞昔布(20 mg/kg,给药前12 h禁食不禁饮),分别于给药前(0 min)和给药后10,20,30,45 min及1,1.5,2,3,4,6,8,12,24 h时眼底静脉丛取血0.5mL,置肝素钠抗凝离心管中,6500×g离心5 min,取血浆-40℃冷冻保存。
待测血浆室温自然解冻,按2.4项下方法对血浆样品进行处理,根据当日随行标准曲线计算每只大鼠各时间点的血药浓度,绘制平均药-时曲线(AUC)。详见图2。采用DAS 2.1.1软件对血药浓度进行统计矩拟合,并计算相关药代动力学参数。详见表2。采用SPSS 13.0统计学软件对两组大鼠的主要药代动力学参数进行统计学分析。结果显示,合用帕罗西汀后,艾瑞昔布的AUC0-t,AUC0-∞,峰浓度(Cmax)均显著升高,清除率(CL)显著降低,差异有统计学意义(P<0.05),表明帕罗西汀可抑制艾瑞昔布的代谢,CYP2D6参与了艾瑞昔布的代谢。
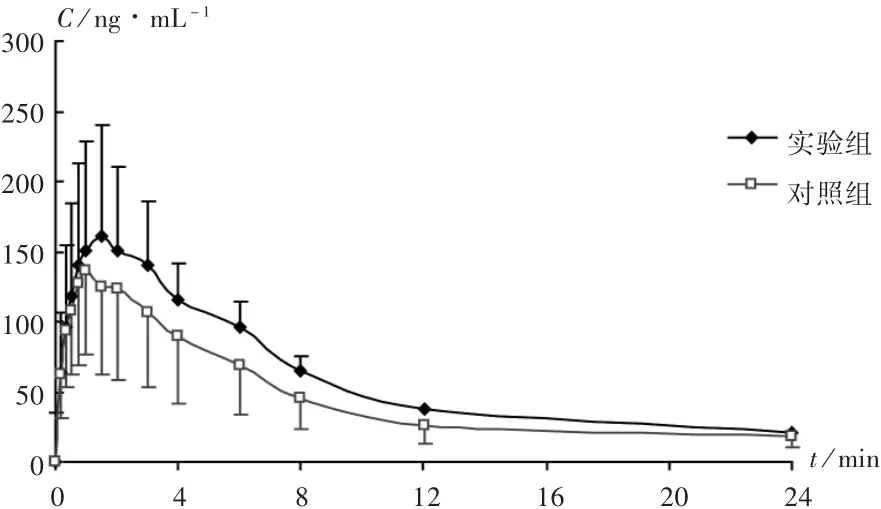
图2 两组的平均药-时曲线(n=18)
3 讨论
CYP2D6在体内的含量仅次于CYP3A,分布于多个器官,如肝脏、大脑和骨骼肌等,代谢20%~25%的药物[11-12]。因此,研究某一药物与CYP2D6的作用关系对于研究该药物的相互作用具有非常重要的意义。目前,只有体外研究结果显示,肝微粒体酶CYP2D6参与艾瑞昔布的体外代谢过程。帕罗西汀为体内CYP2D6的强抑制剂,且帕罗西汀与以CYP2D6酶系为代谢途径的药物联用时,可影响其代谢,升高血药浓度,甚至引起不良反应[13]。故本研究中探讨了CYP2D6是否参与艾瑞昔布的体内代谢过程。
表2 两组大鼠艾瑞昔布药代动力学参数(±s,n=18)

表2 两组大鼠艾瑞昔布药代动力学参数(±s,n=18)
注:t1/2为半衰期,Tmax为达峰时间,CL为清除率,V为表观分布容积。
药代动力学参数AUC0-24h[mg/(h·L)]AUC0-∞[mg/(h·L)]t1/2(h)Tmax(h)CL[L/(h·kg)]V(L/kg)Cmax(mg/L)实验组1 485.5±480.3 1 730.4±606.5 7.3±5.2 1.5±0.7 0.01±0.01 0.12±0.06 192.1±70.8对照组1 125.1±457.6 1 331.3±592.6 7.4±3.8 1.5±0.6 0.02±0.01 0.17±0.07 162.2±53.0 P值0.02 0.05 0.96 0.89 0.03 0.10 0.04
本研究结果显示,与对照组相比,经CYP2D6抑制剂帕罗西汀预处理的大鼠,其体内艾瑞昔布的Cmax增大为原来的1.18倍,表明艾瑞昔布在大鼠体内的吸收速度变快;AUC升高为原来的1.32倍,表明艾瑞昔布在大鼠体内的暴露量增加,生物利用度增加;CL降低为原来的65%,表明艾瑞昔布的代谢减慢。综合分析,帕罗西汀抑制了艾瑞昔布在大鼠体内的代谢,从而使其吸收增加、清除减慢,增大了艾瑞昔布在大鼠体内的生物利用度。考虑帕罗西汀作为CYP2D6的特异性抑制剂,因而推断CYP2D6参与了艾瑞昔布在大鼠体内的代谢。该结果与体外大鼠肝微粒体酶实验[14]和重组人源肝微粒体酶研究[15]所得结果均一致,为CYP2D6参与艾瑞昔布的代谢提供了理论支持。
由于大鼠与人之间存在种属差异,且实验中仅考察了帕罗西汀对艾瑞昔布在大鼠体内代谢的影响,具有一定的局限性。故艾瑞昔布在人体内的代谢途径仍有待进一步研究,以为临床合理用药提供更有力的依据。
本研究中选用帕罗西汀药品而非对照品作为CYP2D6的选择性抑制剂,主要基于以下考虑。首先,在人体的临床研究中,受试者服用的均为药品制剂,为保证和后续临床研究的一致性,从而选择帕罗西汀药品而非对照品;其次,考虑到药用辅料对提高给药对象的安全性及减少不良反应的发生具有一定作用,对实验动物而言,更符合伦理要求;最后,药品辅料属商业机密,获得帕罗西汀的辅料非常困难,且考虑到辅料的安全性已经国家批准,因而对肝酶产生作用的可能性不大。故最终选择帕罗西汀药品作为本研究中的选择性抑制剂。
