Dehydrated patient without clinically evident cause: A case report
2020-04-08FedericaPalladinoMariaCristinaFedeleMariannaCasertanoLauraLiguoriTizianaEspositoStefanoGuarinoEmanueleMiragliadelGiudicePierluigiMarzuillo
Federica Palladino, Maria Cristina Fedele, Marianna Casertano, Laura Liguori, Tiziana Esposito, Stefano Guarino, Emanuele Miraglia del Giudice, Pierluigi Marzuillo
Federica Palladino, Maria Cristina Fedele, Marianna Casertano, Laura Liguori, Tiziana Esposito,Stefano Guarino, Emanuele Miraglia del Giudice, Pierluigi Marzuillo, Department of Woman,Child and of General and Specialized Surgery, Università degli Studi della Campania “Luigi Vanvitelli”, Naples 80138, Italy
Abstract BACKGROUND Patients affected by cystic fibrosis can present with metabolic alkalosis such as Bartter’s syndrome. In this case report we want to underline this differential diagnosis and we aimed focusing on the suspect of cystic fibrosis, also in case of a negative newborn screening.CASE SUMMARY In a hot August –with a mean environmental temperature of 36 °C– an 8-mo-old female patient presented with severe dehydration complicated by hypokalemic metabolic alkalosis, in absence of fever, diarrhea and vomiting. Differential diagnosis between cystic fibrosis and tubulopathies causing metabolic alkalosis(Bartter’s Syndrome) was considered. We started intravenous rehydration with subsequent improvement of clinical conditions and serum electrolytes normalization. We diagnosed a mild form of cystic fibrosis (heterozygous mutations: G126D and F508del in the cystic fibrosis transmembrane conductance regulator gene). The trigger factor of this condition had been heat exposure.CONCLUSION When facing a patient with hypokalemic metabolic alkalosis, cystic fibrosis presenting with Pseudo-Bartter’s syndrome should be considered in the differential diagnosis, even if the newborn screening was negative.
Key Words: Metabolic alkalosis; Dehydration; Cystic fibrosis; Pseudo-Bartter syndrome;Heat exposure; Children; Case report
INTRODUCTION
Cystic fibrosis (CF) is a monogenic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7. CF is complex and greatly variable in clinical expression[1]. Airways, pancreas, male genital system,intestine, liver, bone, and kidney are involved.CFTR-related disorders are conditions determined by mutations in theCFTRgene but not giving the usual CF clinical picture.They are often mild form of CF and their clinical manifestations are limited to a single district and include episodes of recurrent pancreatitis or isolated bilateral bronchiectasis. Males can manifest bilateral agenesis of the vas deferens with no digestive or respiratory involvement[2]. Both CF andCFTR-related disorders can present metabolic alkalosis, such Bartter’s syndrome (BS).
In this case report we want to underline differential diagnosis between cystic fibrosis and tubulopathies causing metabolic alkalosis (such as BS). Moreover, we aimed focusing on the possibility of suspect diagnosis of cystic fibrosis, nevertheless a negative newborn screening.
CASE PRESENTATION
Chief complaints
In a hot August –with a mean environmental temperature of 36 °C– an 8-mo-old female patient come to our observation because of somnolence and reduced response to stimuli.
History of present illness
Weight loss in the last 7 d and low-quantity micturition in the last 24 h were reported.
History of past illness
In the first months of life the patient presented three episodes of upper respiratory tract infections.
Personal and family history
Neonatal screening for cystic fibrosis, hypothyroidism, and phenylketonuria were normal. She was assuming about 400 mL/d of milk. With the exception of 400 IU/d of Vitamin D, no other medications were administered.
Physical examination
She showed slightly dry mucous membranes, tachycardia (140 beats/min) and refill time of about 2 s. Fever was absent as such as vomiting or diarrhea.
Laboratory examinations
Urinalysis did not reveal any abnormality. Serum chemistry was as follows: Venous pH 7.5, bicarbonate 35.1 mmol/L, sodium 135 mEq/L, potassium 2.6 mEq/L, chloride 86 mEq/L, creatinine 0.31 mg/dL, calcium 10.7 mg/dL, phosphorous 4.3 mg/dL,magnesium 2.2 mEq/L. Aspartate and alanine aminotransferase, γ-Glutamyltransferase, glycaemia, bilirubin, erythrocyte sedimentation rate, C-reactive protein,procalcitonin, alkaline phosphatase and complete blood count were within normal limits. Urinary calcium/creatinine ratio was 0.02 mg/mg, fractional excretion of sodium (FeNa) 0.7%. Electrocardiography was normal.
After 36 h of treatment, clinical conditions of the patient and serum electrolytes improved. Venous pH was 7.44, HCO3- 25.5 mmol/L, Na 142 mEq/L, K 4.5 mEq/L,CL 105 mEq/L.
We submitted our patient to sweat test when she became well hydrated with normal acid-base and electrolyte balance. The sweat test showed NaCl in the sweat of 100 mmol/L (normal value < 60 mmol/L). High sweat NaCl values were confirmed at the following sweat test after 2 d (sweat NaCl 87 mmol/L). So, we performed genetics analysis for cystic fibrosis. Molecular diagnosis showed the following heterozygous mutations: G126D and F508del in the geneCFTR(Figure 1). This genotype has been described as cause of mild cases of cystic fibrosis or of atypical forms (better known asCFTR-related disorders)[3].
Imaging examinations
Abdomen ultrasonography was normal.
FINAL DIAGNOSIS
Hypokalemic metabolic alkalosis due to cystic fibrosis presenting with Pseudo-Bartter syndrome.
TREATMENT
When patient was admitted intravenous rehydration with a solution containing NaCl 0.9% and glucose 2.5% with the addition of KCL 40 mEq/m2per day was started.When dehydration status and hypokalemic metabolic alkalosis was resolved this infusion was stopped.
OUTCOME AND FOLLOW-UP
Gene mutation analysis identified two heterozygous mutations of theCFTRgene:G126D and F508del. This genotype has been described as cause of mild cases of cystic fibrosis or of atypical forms (better known asCFTR-related disorders)[3].
The variable phenotype of patient affected byCFTR-Related disorders makes very complicated the genetic counseling. A regular clinical evaluation is necessary because CF symptoms may appear later[1]. She will undergo follow up visits at 3, 6, 12 mo after the diagnosis and yearly thereafter, because the atypical form (orCFTR-related disorders) could get worse over time. Proper immunization and influenza vaccination were recommended[4].
DISCUSSION
The main causes of metabolic alkalosis are shown in the Table 1[5-8]. Our patient had no signs of gastro-intestinal losses neither assumed any drug or too much calcium and absorbable alkali. Therefore, possible causes were or skin (due to cystic fibrosis) or renal (due mainly to BS) losses. However, our patient recovered too promptly compared with a dehydrated patient with BS[9,10]. Moreover, FeNa was < 1%demonstrating extra-renal losses of sodium (Table 2)[11]and increasing the suspect of cystic fibrosis. In the cystic fibrosis, just chloride depletion (in our case through sweating) is the main cause of electrolyte abnormalities. Physical activity, fever, and heat exposure can determinate an excessive sweat production that causes an extracellular fluid volume and chloride depletion.
The extracellular fluid volume depletion leads to a release of antidiuretic hormone with subsequent sodium reduction, and activation of renin-aldosterone system with potassium reduction. Moreover, chloride depletion causes increased renal bicarbonatereabsorption. This mechanism is regulated by a chloride–bicarbonate exchanger(pendrin) located on the intercalated cells, sited in cortical collected duct. When chloride depletion occurs, HCO3-secretion is inhibited by insufficient Cl-for anion exchange. In addition, pendrin is reduced in case of potassium depletion. All these mechanisms determine hypokalemic metabolic alkalosis. According to these pathophysiological mechanisms, chloride supplementation, more than sodium and potassium supplementation, is needed to correct the metabolic alkalosis state. The presence of chloride, in fact, increases pendrin activity, with bicarbonate secretion in the lumen of collected duct[12,13].

Table 1 Main causes of metabolic alkalosis (modified from references[5-8])

Table 2 Bartter’s syndrome and cystic fibrosis: Differential diagnosis (modified from references[9-18])
This kind of clinical and laboratory presentation of CF (known as Pseudo-BS)[14-18],usually, involves children < 2.5 years and is the presenting clinical picture of CF.Episode of vomiting, excessive sweating, heat exposure, fever or respiratory infection could cause a Pseudo-BS, in case of an underlining CF[15,18]. In our case, with the exception of heat exposure, there were no other reasons justifying the dehydration(gastrointestinal fluid losses with diarrhea and/or vomiting, reduction of salt and fluid intake and/or absorption, excessive sweat production for physical activity or fever).
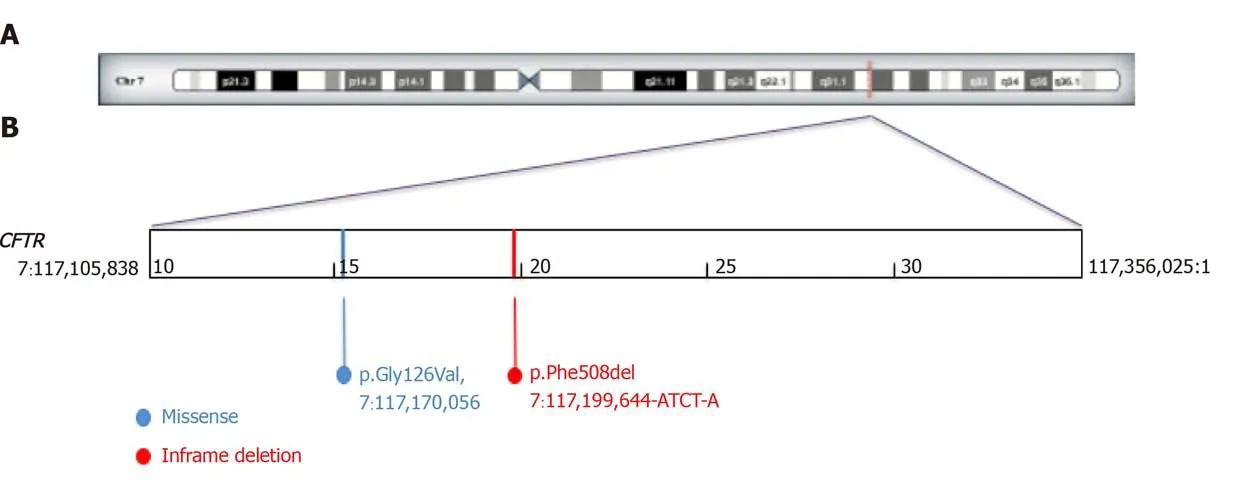
Figure 1 Molecular diagnosis showed the following heterozygous mutations: G126D and F508del in the gene CFTR. A: Locus of cystic fibrosis transmembrane conductance regulator gene on Chromosome 7q31.2. The figure was created and modified by DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembi Resources; B: Graphic view of cystic fibrosis transmembrane conductance regulator with pathogenic variants of our patient.
For this reason, we performed sweat test in the suspect of cystic fibrosis. Sweat test is the gold standard to diagnose classical or atypical forms of CF[19]. In fact, even with over 1000 mutations in theCFTRgene on chromosome 7 are known and it is possible to find children with cystic fibrosis which do not present identifiable gene mutations[20]. Nevertheless, some mutations could show atypical and very mild clinical manifestations[1]. In these cases, sweat test results can be intermediate or negative (2%)and so other diagnostic tests are indicated, if clinical manifestations persist (i.e., geneticCFTRtests,CFTRfunctional tests, such as nasal potential difference, intestinal current measurement)[21,22]. Moreover, it is possible that these patients passed the new-born screening because their sufficient pancreatic status[21-24]. So, clinicians must not exclude,a priori, the suspicious of CF in patients with 1 or more clinical manifestations of CF,despite of negative newborn screening results[25-27].
CONCLUSION
In conclusion, with this case presentation we would highlight that the Pseudo-Bartter’s syndrome could be an initial clinical presentation of cystic fibrosis, and that the heat exposure might be a trigger of this condition. When facing a Pseudo-Bartter’s syndrome, cystic fibrosis should not be excluded from differential diagnosis, even if the new-born screening was negative.
杂志排行

World Journal of Clinical Cases的其它文章
- Relationship between non-alcoholic fatty liver disease and coronary heart disease
- Remission of hepatotoxicity in chronic pulmonary aspergillosis patients after lowering trough concentration of voriconazole
- Endoscopic submucosal dissection as alternative to surgery for complicated gastric heterotopic pancreas
- Observation of the effects of three methods for reducing perineal swelling in children with developmental hip dislocation
- Predictive value of serum cystatin C for risk of mortality in severe and critically ill patients with COVID-19
- Sleep quality of patients with postoperative glioma at home