双离子电池研究进展
2020-04-04周小龙欧学武刘齐荣唐永炳
周小龙,欧学武,刘齐荣,唐永炳
(中国科学院深圳先进技术研究院,功能薄膜材料研究中心,广东 深圳 518055)
1 概 述
1.1 传统电池研究现状
随着能源和环境问题的日益严峻,开发和应用新型高效无污染的能源及储能器件已成为刻不容缓的全球性课题[1-2]。传统锂离子电池(LIBs)由于具有循环寿命长、无记忆效应等特点,现已广泛应用于便携式电子设备、电动工具等领域[3-5]。目前,世界各国已相继提出了大力发展新能源的战略目标,如《中国制造2025》、美国《Battery 500》等。随着锂电池的快速发展,基于锂的电极材料的需求量也急剧增加。据估计,到2050年,锂的总消耗量将达到目前已探明总储量的1/3以上[6]。
从1991年第一款商用LIBs推出以来,为了提高能量密度和安全性,研究人员通过各种手段对LIBs进行了优化和改善[7-8],从最初的钴酸锂(LiCoO2:LCO),到磷酸铁锂(LiFePO4:LFP),再到三元(LiNi1-2xCoxMnxO2:NCM)等正极材料。然而,目前LIBs的电极材料几乎达到理论能量极限[3,9-11]。随着新能源的发展,正极材料所使用的过渡金属(例如钴、镍等)价格快速增长。例如,仅2017年,钴的价格就上涨了145%[12];而丰度相对较少的镍等元素也面临资源枯竭的危险[13]。因此,开发高效低成本的新型储能电池成为下一代储能技术的主要研究方向。其中,不含钴、镍等过渡金属的正极材料已成为研究的热点[3]。同时,由于钠和钾元素在地壳中储量丰富[13-14],且与锂具有相似的电化学性质,因此,基于钠离子[13,15-17]、钾离子[18-19]等电池的开发与利用也成为了当前研究的重要方向。此外,其他新型电池,如:锂-硫[20-22]、碱金属-空气[23-24]、锌-空气[25-26]和双离子电池(DIBs)[11,27-30]等也受到了广泛关注。
在各种新型储能器件中,DIBs由于具有成本低廉、环境友好、工作电压高等特点,逐渐引起了人们的重视[19,27-28,30-31]。DIBs与各类传统电池的比较如表1所示。具体而言,对于传统的石墨-LFP电池,其正极材料LFP约占电池总成本的1/3;而DIBs采用石墨类材料作为正极,使得物料成本大幅降低,尽管其具有明显优势,但是对于DIBs的研究仍处于初期阶段。本文将综述DIBs当前的研究现状和面临的主要挑战,并为进一步深入探索活性离子插层机理,为改善DIBs的电化学性能提出新的理解以及相应的改进策略。
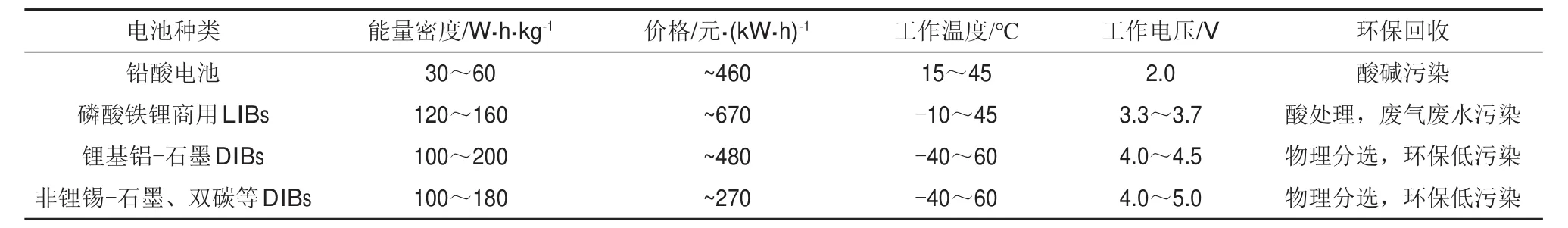
表1 各类电池的能量密度、成本估算、工作电压/温度、环保回收等综合性能比较[12]Table 1 Comprehensive comparison on energy density,cost estimation,work voltage/temperature and recycling property for various types of batteries[12]
1.2 双离子电池的工作原理及特点
如图1所示,DIBs与传统LIBs的电化学工作原理有着显著差异。传统LIBs负极材料通常采用石墨,正极则由含锂的过渡金属氧化物构成。充电时,锂离子从正极脱出,通过电解液插层到石墨负极夹层中,从而将电能转化为化学能;在放电过程中,锂离子从石墨负极脱出,回到正极,将化学能转化为电能[图1(a)]。与LIBs不同,DIBs通常采用石墨类材料作为正极[10-11]。以双石墨-双离子电池为例[图1(b)],充电时,阳离子(如Li+)插层到石墨负极层间,同时阴离子(如PF6-)插层到石墨正极;放电过程则相反,即阴、阳离子分别从正负极脱出回到电解液中。由于石墨正极具有较高的阴离子插层电位(>4.5 Vvs.Li/Li+)[32],使得双离子电池具有高电压的特点,从而有利于获得高的能量密度。此外,石墨正极具有环境友好的特点,并能够有效降低电池的整体成本。
同时,通过上述比较可知,与LIBs中电解液主要起到离子传导的作用不同,电解液作为DIBs活性材料的一部分,对电池的整体性能起着关键作用[27]。即电解液的浓度和阴离子种类对石墨正极的插层电位以及插层容量等都具有重要的影响。在通常情况下,双离子电池体系要求所用隔膜具有较高的气孔率和较大的厚度,以保证电池整个生命周期内电解液充足(图2)。为保证足够的活性离子供应,高浓度电解液将能够有利于减少电解液的用量,降低隔膜的厚度,从而提升电池的稳定性以及能量密度[33]。
另外,由于工作机理的差别,LIBs和DIBs电池容量衰减的主要原因也不尽相同。对于LIBs,电极材料表面SEI膜的不断形成与降解是造成LIBs容量发生不可逆衰减的主要原因。而对于DIBs,其容量衰减主要来自两方面的:其一、电子在负极上的消耗导致阴离子在石墨正极中的不可逆插层,即随着充放电循环的进行,阴离子在充电过程中的“自由空间”逐渐减小,导致容量衰减。其二、阳离子在负极的不可逆插层导致阳离子在负极中可移动的“自由空间”减少,造成容量衰减。简而言之,在初始放电状态,所有的活性离子都储存在电解液中;而在充放电反应过程中,伴随着活性离子往复的嵌入/脱出,电极材料发生的不可逆变化使得活性离子无法全部回到电解液中去,从而导致DIBs性能衰减[33]。
综上所述,DIBs的特点可以归纳为以下几点:①阴、阳离子同时参与电池反应,阴离子插层石墨正极的高电位,有利于提高电池的能量密度;②电解液不仅起到传导离子的作用,同时其作为活性物质的一部分,对DIBs的性能具有重要影响;③电解液作为活性物质使得DIBs对电池隔膜要求高,包括较高的气孔率和保液能力;④活性离子不可逆的插层反应是导致其容量衰减的重要原因。
1.3 双离子电池的发展历程
作为一种新型的储能器件,DIBs经历了一个漫长的发展过程[11,27,29,31,33-36]。图3对DIBs的发展历程进行了简要总结[32,34,37-45]。DIBs的概念是由双石墨电池(DGBs)或双碳电池(DCBs)发展而来,源于石墨插层化合物(GICs)的研究[27,29,31,33,36]。1841年,Schafhäutl[46-47]在硫酸溶液中分析石墨晶片过程中制备出GICs,首次发现阴离子插层石墨的现象。1938年,Rüdorff等[39]证实了阴离子(HSO4-)可逆插层石墨的过程,并以浓硫酸溶液为电解质制备出第一个石墨电池。1989年,McCullough等[48]申请了第一个DGBs电池专利,并提出“双插层”的概念用以解释电池的反应过程,被认为是DIBs发展进程中的里程碑。1994年,Carlin等[49]报道了以室温离子液体为电解液的DGBs,从而开启了阴离子插层石墨作为正极应用的先河。
此后,人们对阴离子插层石墨正极的机理进行了大量研究[42,50-52],包括不同阴离子[30,44,53-55],以及不同溶剂对DIBs性能的改善[28,38,43,56-58]。其中,Seel等[10]对PF6-插层石墨的过程进行了深入的研究。由于阴离子插层石墨正极的电位高(>4.5 Vvs.Li/Li+),使得传统电解液发生分解;而由于大的阴离子半径,阴离子在嵌入/脱出过程中造成石墨容易发生剥落,导致DIBs的循环性能不足。针对以上问题,2012年,Placke等[41]使用具有耐高电压的离子液体电解液,显著提升了双离子电池的循环性能。2014年,Read等制备了基于氟化溶剂和添加剂的高压(5.2 V)电解液体系。2015年,Lin等[43]报道了一种铝离子电池,该电池通过氯铝酸根阴离子(AlCl4-)在石墨正极的嵌入/脱出反应,以及Al2Cl7-阴离子在铝负极的还原氧化反应,实现了电池的充放电。2016年,Tang等[28]报道了一种新型的铝-石墨DIBs,采用廉价金属铝箔同时作为负极活性物质和集流体,使得电池的成本大幅降低。为进一步降低DIBs的成本,近年来陆续开发出基于Na+、K+、Ca2+、Al3+等阳离子的非锂DIBs体系[29-30,36,58]。此外,考虑到实际使用的要求,柔性、全固态、水系等新型DIBs也相继被报道。2018年,Tang等[45]开发出基于聚合物电解质的新型DIBs。2019年,Wang等[34]首次利用转化反应实现了具有良好可逆性的水系新型双离子电池,其能量密度高于目前多数商用正极材料。随后,Sutto等[68]首次提出了反向双离子电池的概念,使得双离子体系得到了进一步的发展。尽管如此,目前DIBs的研究仍处于初级阶段。在接下来的章节中,将具体讨论DIBs中电解液的选择、阴离子插层反应机理、负极的反应机理(例如插层、合金化等),并探讨实现高性能低成本DIBs的有效途径。
1.4 双离子电池电解液的发展
与LIBs类似,DIBs的电解液/质可以是液体、固体、聚合物等[59-61],其中液体或聚合物电解质最为常用[62]。在电解液中,最低未占据分子轨道(LUMO)对应还原电位,最高占据分子轨道(HOMO)对应氧化电位,两者的差值决定了电解液的电压窗口[62]。通常情况下,电池负极费米能级(μu)和正极费米能级(μo)的电化学势应分别与其电解液的LUMO和HOMO能级相匹配。具体而言,μu应比LUMO更低,否则电解液会被还原;而μo应比HOMO更高,以防止电解液的氧化。简而言之,电池正常工作的电压范围应该在电解液的电压窗口以内。传统电解液体系由于能够在负极表面形成固体电解质界面膜(SEI),从而使得电池在高于电压窗口的情况下也能正常工作。尽管如此,由于DIBs中阴离子插层石墨的高电位使得寻找具有高电压的电解液体系显得极为迫切。
1.4.1 有机电解液
作为应用最广泛的电解液类型,研究人员对有机电解液在DIBs中的应用进行了系统研究[55,63-65]。例如,Noel等[55]研究了不同溶剂对阴离子嵌入/脱出的影响。Wang等[63-65]报道了溶剂化对BF4-和PF6-插层石墨行为的影响,包括:碳酸乙烯酯(EC)、碳酸丙烯酯(PC)、丁内酯(GBL)、环丁砜(SL)、碳酸甲乙酯(EMC);并采用非原位和原位等表征手段对阴离子的插层行为进行了系统研究。结果表明,由于EC、GBL和SL对BF4-和PF6-具有较强的亲和性,从而抑制了其在石墨中的插层反应。
1.4.2 离子液体电解液
离子液体具有不易燃、低挥发、高热稳定、宽电化学窗口等优点,在DIBs中的应用前景十分广阔[66-67]。1994年,Carlin等[49]报道了各种室温离子液体作为电解液在双石墨电池中的应用。通过改变EMI+、DMPI+等正离子种类和等负离子种类,构建了基于不同离子体系的双离子电池。例如,以DMPI(AlCl4)作为电解质盐,电池的工作电压为3.5 V,循环效率达到85%。Sutto等[68]结合电化学表征和X射线衍射光谱(XRD),同时采用离子液体电解液,即将咪唑啉阳离子(二取代咪唑啉和三取代咪唑啉)与BF4-、PF6-、TFSI-、双(全氟乙磺酰亚胺)亚胺离子(PFESI-)、硝酸根离子和硫酸氢根离子等阴离子进行配对,研究了各种阴离子在石墨正极中的嵌入/脱出行为。由于阴离子的半径往往大于石墨的层间距,研究人员提出,半径较小的阴离子将更容易进行可逆的插层反应[69]。鉴于此,Meister等[70]研究了不对称氟磺酰亚胺离子插层石墨正极的电化学过程。研究表明,由于FTFSI-阴离子尺寸(0.65 nm)比TFSI-(0.8 nm)小,在相同的充电截止电位下,FTFSI-具有更高的放电比容量。在最近的报道中,研究人员还发现,除了阴离子尺寸以外,阴离子插层石墨的起始电位也取决于电解液,如离子对的形成和自聚集过程等[71]。研究表明,含有不同尺寸的亚胺基阴离子的离子液体电解液,包括BETI-、FSI-、 FTFSI-、 TFSI-、 FSI-/TFSI-, 除尺寸最大的BETI-外,都观察到与阴离子嵌入/脱出相对应的氧化还原峰[图4(a)]。其中,第二大阴离子(TFSI-)的起始插层电位最低,第三大阴离子(FTFSI-)的初始插层电位(4.48 Vvs.Li/Li+)略高于TFSI-(4.44 Vvs.Li/Li+)。虽然FSI-阴离子半径最小,其阴离子嵌入的起始电位反而最大(4.53 Vvs.Li/Li+)。与此同时,两种不同阴离子的混合体系也会影响嵌入/脱出行为[图4(b)],其中混合阴离子的插层电位顺序具体如下:TFSI-/FSI-(4.42 Vvs.Li/Li+) 不同于传统“摇椅”式锂离子电池,DIBs的容量主要依靠阴离子插层正极实现。因此,深入了解阴离子插层机理,对进一步提高DIBs的性能具有重要意义[72]。石墨具有独特的ABA型六方碳环构成的层状结构,层间相互作用较弱,因此,能够允许充/放电过程中阴离子的嵌入/脱出。阴离子嵌入石墨层间的结构特征主要取决于活性阴离子本身的特性,同时,阴离子间复杂的静电和弹性相互作用也会影响后续阴离子的插层动力学。阴离子插层石墨正极的过程可以由相邻的阴离子嵌入层之间石墨烯的层数来描述[46]。通常,用“stage”来定义在无缺陷石墨晶格中由于阴离子插层所形成连续相的状态[73],以及不同“stage”阴离子插层石墨正极的结构[41,74]。关于阴离子插层石墨正极的机理已有较多报道[46,56,65,75],所涉及的阴离子包括:六氟磷酸根离子(PF6-)[10,58,76]、四氯铝酸根离子(AlCl4-)[56,77]、 双(三氟甲磺酰)酰亚胺离子(TFSI-)[42,71,78]、氟磺酰基-(三氟甲磺酰基)酰亚胺离子(FTFSI-)[70-71]、高氯酸根离子(ClO4-)[50,79]、双(氟磺酰基)酰亚胺离子(FSI-)[71,80]、四氟硼酸根离子(BF4-)[63]、三(五氟乙基)三氟磷酸根离子[(C2F5)3PF3-][81]、双(全氟-乙基磺酰基)亚胺离子(BETI-)[71]、三氟甲磺酸根离子(CF3SO3-)[81]、二氟(草酸)硼酸根离子(DFOB-)[80]、四氟铝酸根离子(AlF4-)[82],其结构如图5所示。其中,PF6-与TFSI-为最常见的插层阴离子,接下来将详细介绍。 2.1.1 PF6-离子的插层机理 基于PF6-阴离子的电解液广泛应用于DIBs中。Seel等[10]通过原位XRD表征发现,在充/放电过程中,PF6-在石墨中的电化学嵌入/脱出过程对应着一系列可逆的“stage”相转变。Read等[83]研究了高定向热解石墨在发生PF6-阴离子插层过程中的相变和晶格膨胀现象。采用原位XRD和膨胀测定法,观察到阴离子插层过程中的不同“stage”相的形成和转变如下:C24PF6(stage IV)→ C24PF6(stage III)→ C24PF6(stage II) → C20PF6(stage II) → C24PF6(stage I)→ C20PF6(stage I)。Li等[84]采用原位光学平台实时监测石墨正极在充/放电过程中的厚度演变,发现石墨正极的厚度在最初时呈现出相当大的变化。理论结合实验,Miyoshi等[37]研究了PF6-嵌入石墨中的特性,给出了其插层石墨层中的优化几何形状,并得出PF6-阴离子的理论扩散活化能约为0.23 eV。由于其较大的离子尺寸,PF6-阴离子优先沿着(100)方向扩散;随着PF6-插层石墨程度的增加,其扩散系数相应降低。 2.1.2 TFSI-离子的插层机理 离子液体由于具有蒸气压低、化学稳定性好、电化学窗口宽等优点而备受关注[75]。Placke等[41]采用离子液体电解液,对TFSI-阴离子在石墨正极中的插层机理进行了详细研究[42,74]。结果表明,TFSI-阴离子嵌入后,石墨出现了分级现象。通过电化学原位XRD表征GICs的特定“stage”相转变过程,在充放电过程中观察到XRD峰位的分裂和偏移现象。TFSI-阴离子插层石墨的平均面间距di和相应的间距变化Δd分别为(7.99±0.04)和(4.64±0.04)Å,对应于其体积膨胀约139%。 2.1.3 其他类型阴离子及其插层动力学 考虑到不同阴离子的不同插层效应,Placke等[41]系统研究了一系列阴离子的石墨插层行为,包括PF6-,BF4-,TFSI-和 FTFSI-等。Beltrop 等[71]通过详细研究不同亚胺基离子液体电解液插层石墨正极的行为,阐明了电化学性能与阴离子大小的关系。其中,阴离子嵌入石墨的初始电位有如下关系:BETI->FSI->FTFSI->FSI-/TFSI->TFSI->TFSI-/FSI-。Tasaki等的研究则表明,阴离子插层石墨的能量势垒与石墨和插层阴离子(如PF6-和ClO4-)之间的电子转移密切相关。此外,Wang等[85]利用复合阴离子(PF6-和AlF4-)插层协同效应,设计了一种新型的铝-石墨电池。 由于石墨正极材料的活性位点有限,以及阴离子的离子半径较大与石墨的层间距不匹配等原因,DIBs正极材料面临着比容量不足、倍率性能较差等问题。因此,为了提高正极的性能,目前主要采取两种策略:①对材料结构进行优化;②开发新型正极材料。前者主要包括对具有不同结构的碳质正极的设计、石墨的结构调控、石墨正极的掺杂和缺陷调控。后者旨在突破石墨类正极比容量的瓶颈,开发更具潜力的阴离子宿主材料。 2.2.1 纳米结构设计 众所周知,纳米化可以提高电极材料的比表面积,缩短离子扩散的距离,从而提高动力学性能。因此,石墨正极的纳米化有望提高阴离子在石墨正极中的插层动力学,从而改善石墨正极的电化学性能[86]。Dai等[43,87-88]比较了 AlCl4-阴离子在不同类型石墨正极的插层行为,包括热解石墨(PG)和三维石墨泡沫(3DGF)[图6(a)、(b)]。与PG相比,多孔的3DGF在垂直方向允许石墨烯层与电解质充分接触,缩短了AlCl4-阴离子的扩散路径,从而显著提升了倍率性能。另外,为了提高比容量,Yu等[89]基于等离子体刻蚀工艺开发了一种由石墨烯纳米带(GNHPG)组成的多孔3DGF正极材料[图6(c)]。丰富的纳米空隙为整个GNHPG提供了丰富的阴离子扩散通道,促进了AlCl4-阴离子的嵌入/脱出动力学,同时增加的可容纳AlCl4-阴离子的活性位点,有效提高了石墨正极的比容量[图6(d)]。 2.2.2 掺杂和缺陷调控 碳质正极材料的比容量取决于能够容纳插层阴离子的活性位点的数量,而阴离子在层间的扩散行为也与活性和非活性位点有很大关系。增加活性位点可以提高碳质正极的比容量,减少甚至消除非活性位点有利于增强阴离子的扩散动力学性能。同时,正极材料中的缺陷和掺杂对其电导率起着至关重要的作用,从而影响阴离子的反应动力学。因此,缺陷调控和掺杂设计是提高阴离子插层动力学的一种重要策略。 Chen等[90]在3000℃退火工艺下制备出一种具有高度结晶性、无缺陷的少层石墨烯气凝胶(GA-3000)正极。与具有大量缺陷的石墨烯气凝胶正极相比,无缺陷多孔结构型正极具有高的放电比容量(约 100 mA·h·g-1)。此外,循环 25000次后,容量保持率达到97%,且具有良好的倍率性能。拉曼光谱表明,在AlCl4-阴离子电化学嵌入/脱出过程中,GA-3000几乎没有呈现出缺陷结构。该设计显著降低了插层势垒,促进了AlCl4-阴离子的插层动力学。对比研究表明,缺陷部位不能作为容纳AlCl4-阴离子的活性位点,且缺陷的存在会阻碍AlCl4-阴离子的嵌入/脱出,并降低石墨正极的电导率。因此,对于石墨正极材料的设计应该遵循的原则是:缺陷越少,电化学性能较好。另外,研究表明,适量的掺杂是提高电极电化学性能的重要因素。例如,氮掺杂能够产生更多的活性位点,促进阴离子的插层动力学[91]。Lu等[92]对低成本微孔硬碳正极材料进行了氮掺杂改性,一方面有效改善了循环过程中阴离子反复嵌入/脱出导致的石墨片层剥落问题,并缓解了充放电过程中由体积膨胀引起的应力;另一方面通过掺杂为阴离子吸附提供更多的活性位点,有利于增强阴离子的吸/脱附动力学。因此,氮掺杂微孔硬碳(NPHC)在充放电循环前后可以保持相对稳定的结构,呈现出良好的倍率性能以及较高的比容量。 2.2.3 新型正极材料 有机材料作为正极材料在DIBs中已经得到了广泛的关注[95-96]。例如,Rodriguez-Perez等[93]报道了Coronene结构的多环芳烃(PAH)正极[图7(a)],发现该有机材料具有可逆的阴离子存储特性。研究表明,在阴离子插层反应过程中,(002)和(111)双峰同时向低角度移动,并诱发材料体积膨胀,降低PAH正极材料的结晶度。在放电过程中,双峰恢复到原来位置,表明PAH结构可以恢复到原来的状态。同时,开发无锂体系是低成本DIBs的重要策略。Fan等[94]则将有机PTPAn作为钾基DIBs的正极。发现,对于PTPAn正极,充电后,PF6-阴离子插层并与N+位点相互作用,产生了N+PF6-的亚稳态键合[图 7(b)][94]。该 DIB 在50 mA·g-1时放电比容量为 60 mA·h·g-1。近来,金属-有机框架材料(MOFs)由于其独特的多孔结构而受到关注,其阴离子迁移通道宽、路径短,理论上有利于增强阴离子插层动力学[97]。总的来说,有机材料正极的研究正逐渐受到关注,已有部分有机材料在DIBs中被详细研究。 负极的反应动力学对DIBs的电化学性能同样具有重要影响。负极材料的反应机理同传统LIBs相似,包括插层、合金化、吸附、转化和剥离/沉积等。鉴于插层与合金化过程是大多数DIBs负极最主要的两种反应机理,本节重点介绍这两种机制。 3.1.1 插层机理 石墨作为一种典型的负极插层材料,锂离子Li+插层石墨的反应如下 该反应最显著的特征是分级现象LixC6(0 3.1.2 合金化机理 合金材料(例如:Al、Sn、Si、Zn、Bi等)由于其高的理论容量,导电性好,储量丰富,环境友好等优点,是极具发展前景的负极材料[102-103]。特别是铝和锡等已在DIBs负极材料中得到详细研究。然而,如何解决合金化负极充放电过程中体积膨胀导致的粉化问题是其应用的关键[28-30,36,43,104-107]。本节将对铝和锡负极的合金化反应机理进行系统讨论。 3.1.2.1 铝金属合金化机理 Al作为负极材料主要有以下几个优点:(i)高理论比容量(LiAl约为993 mA·h·g-1);(ii)导电性优异;(iii)成本低廉[108]。根据Li-Al二元相图[图9(a)],室温下锂铝合金可以形成3种稳定的化合物:LiAl、Li3Al2和 Li9Al4。以 Li9Al4为例[109],其理论容量可以高达2235 mA·h·g-1。然而,Al负极的膨胀粉化问题仍然是其所面临的一个巨大挑战。早期研究中,在高温条件下Li-Al合金的电化学反应有所涉及。如图9(b)所示,Wen等[110]在432℃条件下测得了Li-Al系统的库仑滴定曲线。其中,两相3个区域(α+β、β+γ、γ+L)形成在嵌锂的3个不同的平台,3个倾斜的区域也观察到相应的3个单相区域(α、β、γ),形成的金属间化合物与锂铝二元相图吻合良好[109]。然而,在室温下,Li-Al系统的电压剖面仅在第一次锂化过程中表现出0.26 V的一个宽平台[图9(c)][111]。初始电压区域I(2~0.26 V)与铝电极顶部氧化层的存在有关。电压平台(区域II)被指定为LiAl相的形成,产生包括Al和LiAl的两相区域。区域III(0.26~0.01 V)的倾斜特征表明没有形成其他富锂相(如Li9Al4)。 此外,在第一个充放电周期内还观察到较大的不可逆容量损失,可能是由于铝电极在一个完整的周期后出现微裂纹失去电连接所致[112]。Tang等[44]对满充态铝负极的物相分析发现:铝-石墨DIBs中铝负极的反应产物为AlLi相[图9(d)]。为了更好地了解反应动力学,不同学者对锂在AlLi相中的化学扩散进行了研究。由于测量过程中使用的电极材料的择优取向不同,锂在AlLi相中的化学扩散系数从1×10-9~1×10-12cm2·s-1之间变化[111,113]。例如,使用火法冶金得到的AlLi合金作为各向异性的负极材料(β-LiAl),在(24±2)℃的条件下,Li+在其中的扩散系数约为(7×10-9±3×10-9)cm2·s-1[114]。Kumagai等[115]选用沿(100)择优取向的β-LiAl作为负极,得到的扩散系数值为1×10-10cm2·s-1。在20℃条件下[114],无择优取向的非晶态LiAl相的化学扩散系数约为6×10-12cm2·s-1。另外,扩散系数也与材料的有效表面积以及表征方法有关。 3.1.2.2 锡金属合金化机理 与Al相比较,Sn具有更高的活性,能与多种非锂活性阳离子反应形成合金,如Na+、K+、Ca2+[29,36]。为了更好地了解Sn负极,本节将简要概述Na-Sn、K-Sn和Ca-Sn体系的电化学反应机理。 Na-Sn体系:在室温条件下,Na-Sn可以形成7个中间相 ,即 : NaSn6、 NaSn4、 NaSn3、 NaSn2、Na9Sn4、Na3Sn和 Na15Sn4[116]。而在120℃时,可以观察到3种Na-Sn合金:NaxSn-I(1.0≤x≤1.25),NaxSn-II(2.25≤x≤2.5)和NaxSn-III(3.5≤x≤3.75)。这些实验结果与DFT计算结果一致,但NaSn5相则超出了Crouch-Baker等[117-118]所给出的成分范围。在30℃下[119],虽然得到的充放电电压平台与DFT结果吻合较好,但前3个平台末端形成的化合物与Na-Sn相图中已知的相并不相同。其中,Komaba等[120]通过实验证明,在第4个平台末端形成的化合物是Na15Sn4。以磁控溅射法所制备的Sn膜作为电极参与电池反应,通过对比Na-Sn平衡相图可以得到Na5Sn4合金相,且此相为亚稳态相[121]。根据Tang等[30]的报道,在钠基DIBs中Sn负极的最终产物为NaSn相。虽然NaSn相的形成使Sn负极(约120%)的体积膨胀比Na15Sn4(约420%)小得多,但其结构不稳定。 K-Sn 体系:在室温条件下,K-Sn平衡相图中呈现5个中间相,即K4Sn23、KSn2、K2Sn3、KSn和K2Sn[122]。Sultana等[123]在 KIBs中采用 Sn-碳复合负极,从实验上证实了Sn与K的合金化反应机理。在充分放电后,通过XRD分析,产物被认定为K2Sn5和K4Sn23。虽然K2Sn5的形成目前还需要进一步鉴定,但KSn合金相已被多个小组证明是KIBs的最终产物[124-125]。此外,相比于初始Sn箔,钾基DIB(Sn||膨胀石墨)的充电状态下,在Sn负极形成了不同的合金产物[36],且其XRD结果显示明显的K2Sn相峰。K2Sn相是K-最富相,最高的理论容量为452 mA·h·g-1。然而,其相对较大的体积膨胀(约360%)使得Sn箔的循环稳定性较差。 Ca-Sn 体系:在室温条件下,Ca-Sn平衡相图中有7种不同中间相[图10(a)],包括CaSn3、CaSn、 Ca7Sn6、 Ca31Sn20、 Ca36Sn23、 Ca5Sn3和Ca2Sn[126]。到目前为止,对于Ca-Sn体系的电化学反应机理的报道还较少。Lipson等[127]报道了一种以六氰酸锰为正极,锡为负极的钙离子电池,但并没有具体阐明负极的反应机理。Tang等[29]报道了Sn箔为负极,石墨为正极的钙基DIB,系统分析了Ca-Sn体系的电化学反应机理,证明其电化学反应的最终产物为Ca7Sn6相(图10b)。如图10(c)~(e)所示,在充电至4.0 V时,Ca7Sn6的(201)峰出现在22.8°附近;伴随着充电过程的持续进行,(201)峰值强度逐渐增加,并在充电结束时(5.0 V)达到最大值。在随后的放电过程中,峰值强度逐渐降低,在3.0 V时消失,表明合金化过程具有良好的可逆性。DFT计算表明,不同环境下Ca7Sn6晶格中,Ca—Sn键的形成能均为负值,表明Ca7Sn6相热力学形成的可行性。 DIBs负极材料仍存在理论容量低、插层负极材料反应动力学缓慢以及基于合金化反应的负极材料体积膨胀等问题。值得注意的是,插层和合金化机制在锂电池负极中也普遍存在[128],因此,已报道的改性策略对DIBs负极材料的改性具有一定的借鉴作用[11,27-31,33,128]。本节重点阐述用于DIBs的插层和合金化类负极材料的研究进展和针对上述问题提出的改进策略。 3.2.1 负极材料的插层反应的改进策略 多孔结构设计:插层负极理论容量低、反应动力学差,限制了其能量密度和倍率性能的提高。为了解决这些关键性问题,研究人员提出了多孔结构设计改性策略,其优点如下[129-130]:①电解质能够充分与电极表面接触;②大表面积促进电极/电解质界面上的电荷转移;③缩短离子扩散路径;④提供快速电子通道;⑤提供空隙空间来适应体积变化,从而缓解活性材料粉化的问题。基于上述多孔结构的优点,合成出具有多孔结构的软碳(SC)作为负极[44],其更大的层间距离(SC的0.39 nm大于石墨的0.34 nm)有利于Na+的插层,提升倍率性能。一方面,交联多孔结构将为电解液提供进入SC内部的良好通道,并能有效缓解钠化过程中的体积变化。同时,三维骨架结构提供了一个稳定的框架和快速的电子传输途径。此外,该结构具有较宽的条纹厚度(30~60 nm),有利于缩短Na+扩散长度。采用该多孔SC作为钠基DIB负极,获得了良好的电化学性能。 复合材料设计:碳包覆复合材料设计也可以有效改进DIB负极[131-132]。其优点如下[133-135]:①改善离子或电子传导性;②促进固体电解质界面(SEI)层的形成;③提高结构稳定性。例如,二硫化钼(MoS2)具有670 mA·h·g-1的理论比容量,作为钠基DIBs的插层负极材料具有广阔的应用前景[136]。然而,MoS2的导电性较差。通过碳层包覆可以有效提高其导电性,降低电荷转移电阻。Zhu等[131]开发了一种通心粉状MoS2/C纳米复合材料。该纳米管结构的平均长度、内部直径和壁厚分别为1.5 μm、200 nm和100 nm。同时,其层间距约为0.98 nm,比石墨(0.34 nm)大得多,从而有利于Na+的嵌入/脱出。基于该MoS2/C纳米复合材料为负极,石墨为正极的钠基DIB(MoS2/C-G DIB)表现出良好的倍率性能。 3.2.2 合金化负极改进策略 体积膨胀大、循环性能和倍率性能差是合金化负极材料的主要挑战。例如,Sheng等[30]报道了一种基于Sn箔负极的钠离子DIB,循环400次后容量保持率94%。而采用Sn作为负极的钾基和钙基DIBs的循环寿命不足400次[30,36]。因此,要实现基于合金化负极的低成本、高性能DIBs的开发,首先必须解决负极材料体积膨胀粉化的问题。目前,对负极材料(如Sn)进行改性使其能够与低成本阳离子(Na+、K+、Ca2+等)实现可逆合金化并应用于DIBs的研究报道还较少。本节重点介绍合金负极材料(以Al为例)用于锂基DIBs的策略,包括多孔结构设计、碳包覆复合材料、界面改性、纳米结构设计等。 多孔结构设计与碳包覆复合材料:Jiang等[105]报道了一种一体化结构设计策略改性Al负极。该设计中[图11(a)],在三维多孔玻璃纤维隔膜一侧直接沉积Al膜,构建具有多孔结构的Al负极,并采用天然石墨作为正极[图11(b)]。在铝锂合金化过程中,该多孔结构能够容纳体积膨胀,而玻璃纤维提供了一个坚固的支架,保证铝负极的结构完整性。此外,多孔结构也增加了铝负极与电解液之间的接触面积,促进了界面电荷转移。这种一体化结构的DIBs,显示出优异的长循环稳定性。在60 C的高倍率条件下1500次循环后仍有约61%容量保持率,且多孔铝负极结构保持完整,没有明显粉化。另外,碳包覆结构也是提高电极材料电化学性能的另一种有效策略。Tong等[107]将多孔结构与碳包覆复合材料设计相结合,开发了一种用于锂基DIBs的碳包覆多孔铝箔(pAl/C)负极。如图11(c)所示,pAl/C负极具有碳层均匀涂覆的毛孔状结构(直径约1 μm和长度约20 μm)[图11(d)]。由于多孔结构中离子扩散距离的缩短,显著提高了其倍率性能。 纳米结构设计:与正极材料类似,纳米结构设计也适用于负极材料的改性。例如,为了进一步提高锂基DIBs在高倍率下的循环稳定性,开发出基于碳包覆铝纳米球(nAl@C)的负极材料[137]。铝纳米球(直径50~100 nm)被一层约5 nm的薄碳层包覆[图11(e)~(g)]。基于nAl@C的DIBs,在15 C倍率下充放电1000次循环后,放电容量仍保持88 mA·h·g-1。说明碳包覆纳米球能够有效缓解充/放电过程中的机械应变和应力。 界面改性:电极界面性质(组成、结构、形貌等)在电化学反应中起着重要作用。Zhang等[138]设计了一种三维多孔聚合物层修饰的铝负极,紧密的界面接触可以防止铝负极在反复合金和去合金化过程中发生严重的粉化和表面裂纹。此外,三维多孔聚合物层具有较高的电解质吸收率和优异的离子导电性。在2 C的电流密度下,锂基DIBs具有良好的循环稳定性。1000次循环后,容量保持率为92.4%。此外,Qin等[106]开发了一种新型铝负极,即在铝负极表面构建了一层铝-碳中空界面结构。该设计能有效地通过空心结构释放应力,缓冲合金化过程中的体积膨胀,其长循环寿命达到1500次以上,且容量保持率高达99%。 3.2.3 探索新型负极材料 除了上述负极材料的结构设计和改进外,开发新型负极材料也是一种极具潜力的改进策略,如过渡金属氧化物(TiO2、MoO3、Nb2O5等)[139-141]、过渡金属盐(FePO4)[142]、二维层状金属硫族元素化物 、 MOFs[143-144]、 MXenes (Ti3C2、 Ti2C、 V2C、Nb2C、Ti3CN等)[145]和其他新型碳基负极材料[146]等。 过渡金属氧化物及其盐:Yoshio等[139-141]报道了一系列过渡金属氧化物作为DIBs的负极材料,如TiO2、MoO3、Nb2O5。例如,采用 Nb2O5负 极的DIBs具有较好的电化学性能,循环50圈容量保持率仍在90%左右[140]。除了过渡金属氧化物以外,过渡金属盐也具有作为DIBs负极材料的巨大潜力。Dong等[147]制备了一种新型的还原氧化石墨烯包覆的Na2Ti3O7负极材料(NTO@G)。该NTO@G由宽50~200 nm的NTO纳米线和层间距为0.84 nm的半透明石墨烯层复合而成。采用NTO@G作为钠基DIBs负极,5000次循环后,仍保留80%的容量,循环稳定性良好。 2D层状材料:除了过渡金属氧化物及其盐,各种层状二维材料也具有巨大的吸引力。例如,金属硫族化合物,如SnS2、WS2、CoS2、和2D-MOFs,如Mn-、Ni-、Co-2D MOF纳米薄片[150-151]已经被逐步研究应用于LIBs和/或KIBs。如MXenes,作为一种独特结构的新型层状材料,其在被Gogotsi等[152-153]发现以后就获得了广泛关注。作为LIBs、NIBs、KIBs的负极材料,其理论比容量分别可以达到447.8、351.8、191.8 mA·h·g-1[154-155]。 高效低成本并且环保的新型储能技术在未来储能领域具有广泛的应用前景。DIBs作为一种具有电压高、成本低、环境友好的新型储能器件,最近受到了极大关注。同时,由于传统LIBs面临资源短缺、成本上升等问题,大力发展地壳丰度高的低成本金属离子(如Na+、K+、Ca2+、Al3+等)基DIBs已成为当前研究的一个热点。本文概述了DIBs的发展历程、电池结构及反应机理,分析了DIBs中涉及的反应动力学,包括电解液体系,正极的阴离子插层机理、负极的阳离子插层与合金化动力学等。针对DIBs存在的问题,从电解液选择、电极材料结构设计以及新型电极材料的开发等方面论述了近年来低成本高性能DIBs的研究进展和改进策略。尽管DIBs的研究已取得良好进展,但仍然面临诸多挑战,如:①比容量和能量密度不高;②非锂阳离子反应动力学相对较差;③低成本电解质盐溶解度较低;④库仑效率不高;⑤缺乏对反应动力学的深入理解等。可以预见,通过克服上述关键问题,DIBs有望实现在储能电站、低速电动车、基站储能等领域的实际应用。2 DIBs正极的反应机理及其改进策略
2.1 正极的反应机理:阴离子插层
2.2 正极反应动力学的改进策略
3 DIBs负极的反应机理及其改进策略
3.1 负极的反应机理
3.2 负极反应动力学的改进策略
4 总结与展望
