密度泛函理论研究SiFCl自由基和异硫氰酸的反应机理
2020-03-30侯丽杰韩彦霞高立国
侯丽杰,韩彦霞,高立国
(1.陇东学院 化学化工学院,甘肃 庆阳 745000;2.榆林学院 化学化工学院,陕西 榆林 719000)
异硫氰酸(HNCS)是众所周知的异氰酸(HNCO)的等电子体[1,2]。在含硫燃料燃烧过程中,HNCS及其派生自由基NCS的重要中间体,参与迅速除去燃烧的废气中有毒NO4化合物的过程[3-5],所以各国学者十分关注异硫氰酸与小分子自由基的反应[6-18]。Sun[18]等利用正则变分过渡态理论的动力学方法研究了HNCS与CN自由基的反应机理,结果表明C进攻S生成HCN和NCS的反应为主反应通道。硅氟自由基作为许多硅氟化合反应重要的中间体,由于有较强的反应活性,与HNCS的反应是个令人感兴趣的课题,但由于自由基反应的复杂性使得此类反应的机理及相关信息很难从实验上获得。本课题组在对HNCS与SiF2和SiHF自由基的反应机理研究的基础上[16,17],采用泛函密度理论(DFT)的B3LYP方法,讨论HNCS与SiFCl自由基的反应机理,力求从理论上揭示其反应特征。
1 计算方法
使用DFT的B3LYP方法[19,20],以6-311++G**为基组,并把反应势能面上的所有稳定点(反应物、过渡态、中间体)进行了全几何参数优化,并通过简谐振动频率分析确认过渡态的正确性,反应通道中反应物、中间体、过渡态及产物的连接关系通过IRC的计算得以验证。为了获得更精确的势能面能量信息,各反应通道上驻点的单点能又用G3方法进行校正,并依据此结果,以反应物作为能量零点计算了各驻点的相对能量,并得出反应能级图。所有计算均在Gaussian 03程序上完成[21]。根据Winger校正的Eyring过渡态和统计热力学理论,利用自编程序,对不同温度下低势垒反应的平衡常数和速率常数进行计算。为了进一步验证异硫氰酸和SiFCl自由基的反应机理,又在B3LYP/6-311++G**水平上对反应入口处的构型进行自然键轨道理论(NBO)[22]和电子密度拓扑分析(AIM)[23]。
2 结果与讨论
2.1 初始中间体的形成
计算结果表明,SiFCl自由基单重态能量比三重态能量低262.05kJ·mol-1,所以本文只研究单重态的SiFCl自由基和异硫氰酸的反应。NBO计算结果表明,在单重态下的SiFCl自由基与HNCS接近时,HNCS中N的孤对电子与SiFCl中Si原子中空p轨道相互作用形成反应物前驱络合物IM6,削弱了N-H键,因此,从IM6发生H迁移反应。由于HNCS中S的孤对电子分别与SiFCl自由基中Si原子中空p轨道和σ*(Si-Cl)作用形成IM4和IM5,开始发生抽提S的反应。HNCS中的S的孤对电子与Si原子的空p轨道相互作用分别经过渡态TS1和TS2形成IM2和IM3,在IM2中,σ*(Si-N)和σ*(Si-C)和F中的孤对电子相互作用使C-N键削弱,发生了抽提NH反应,所以,SiFCl自由基与HNCS的反应是复杂的多通道反应。
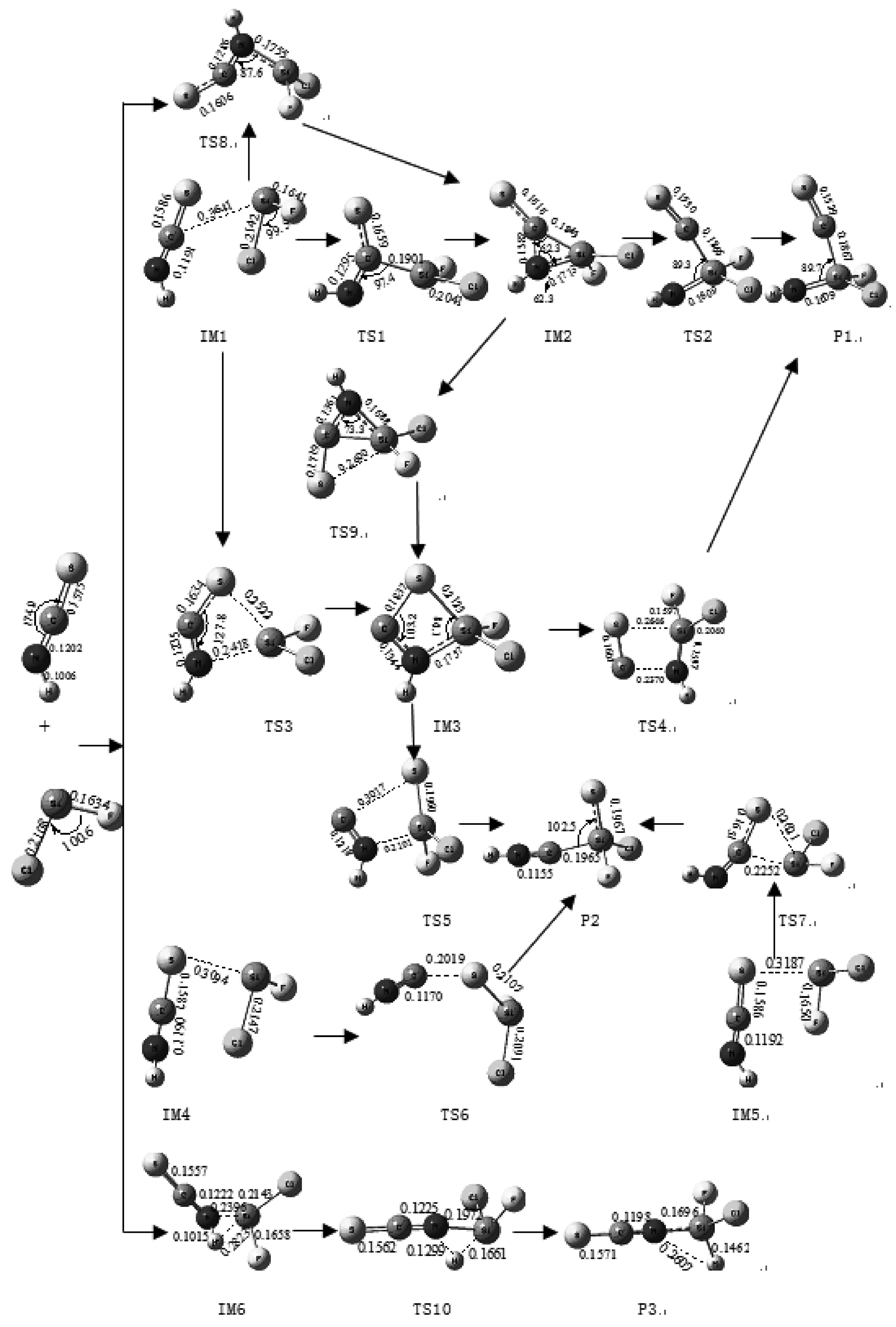
图1 反应通道中各物种的几何构型示意图(键长/nm;键角/度)
2.2 反应路径分析
图1为可能反应通道中各物种的几何构型示意图,图2是在G3/B3LYP/6-311++G**水平下反应的势能面图〔以反应物为零点的相对能Erel(kJ·mol-1)〕。表1列出反应通道入口处主要化学键的电子密度拓扑性质,表2为1atm下,在100-1000K温度范围内,反应的平衡常数K和反应速率常数k(s)的值。以下将分别对各反应通道进行分析讨论。
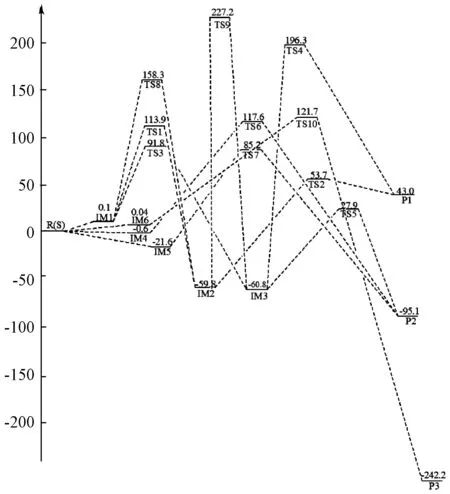
图2 G3/B3LYP/6-311++G**水平下反应的势能面图(相对能量单位kJ·mol-1)
2.2.1 抽提S反应
由图2可以看出,HNCS和SiFCl自由基靠近形成IM4和IM5,能量分别降低0.63kJ·mol-1和21.6kJ·mol-1。IM4和IM5分别经过渡态TS6和TS7生成产物SSiFClCNH,其反应势垒分别为118.2kJ·mol-1和106.8kJ·mol-1。IM4→TS6→P2,Si-S键长缩短(由0.3094nm至0.1967nm)。而且由表2可知,Si-S键成键临界点(BCP)的电荷密度ρ(r)从0.0147逐渐增加至0.1297。IM5→TS7→P2,Si-S键长缩短(从0.3187nm到0.1967nm),Si-S键BCP的ρ(r)从0.0157逐渐增加至0.1297,且▽2ρ(r)值逐渐增加。这表明Si-S键强度逐渐增加,直至最后成键,Si-S键离子性逐渐增强。椭圆度ε逐渐减小,也表明Si-S键的π键特性减弱,表现为σ键特性。
由图2和表2可以得知,生成P2的反应放热95.1kJ·mol-1,且在100-600K温度区间内,反应的平衡常数均较大,但IM5经TS7生成P2的速率常数比IM4经TS6生成P2大。因此,IM5→TS7→SSiFClCNH(P2)反应为主反应通道。
由图2可知,IM1经TS3形成四元环中间体IM3,而IM3经TS5生成产物P2,反应势垒为88.7kJ·mol-1。由图1可知,Si-N键长从0.1757nm(IM3)拉伸至0.2101nm(TS5),Si-S键长从0.2123nm(IM3)缩短至0.1967nm(P2),有利于Si-N的断裂和Si-S的生成。由图2可知,IM1经TS8形成三元环中间体IM2,反应势垒为158.2kJ·mol-1;IM2经TS9形成四元环中间体IM3,反应势垒为286.7kJ·mol-1。因此,由于反应势垒较高,以上三条经三元环和四元环抽提S反应通道都不是主反应通道。
2.2.2 H迁移反应
SiFCl自由基和HNCS相互作用生成反应物中间体IM6,IM6经TS10生成(HSiFClNCS)P3。由图1所示,N-H键长从0.1015nm伸长至0.2602nm。在反应过程中,Si-H和Si-N键长分别从0.1661nm、0.1972nm缩短至0.1462nm、0.1669nm。表明N-H键被削弱,逐渐断裂,Si-H键,逐渐生成的过程,HNCS中的H原子转移至SiFCl自由基的Si原子上,完成了H转移反应。由表1可知,Si-H键BCP的(ρ(r))逐渐增加,表明键强度增加,直至成键。▽2ρ(r)值从负值[-0.1046(TS10)]逐渐变为正值[0.2170(P3)],表明Si-H键由共价性逐渐变为离子性。Si-H键的ε逐渐降低至0.0203(P3),表明Si-H键π键特性减弱,表现为σ键特性。在反应过程中,Si-N键BCP的ρ(r)从0.0381(IM6)逐渐增加至0.1270(P3),表明键强度逐渐增强,直至最后成键。Si-N键的▽2ρ(r)值逐渐升高,表明键的离子性逐渐增强。
如图2所示,IM6经TS10生成P3,其反应势垒为46.8kJ·mol-1,整个反应放热242.2kJ·mol-1。此反应通道不管在热力学还是在动力学上都是有利的。但从表2可知,在100-1000K范围内,反应的平衡常数和速率常数都很小,这可能是由于HNCS和SiFCl自由基相互作用形成中间体IM6时,能量升高74.9kJ·mol-1。因而H转移通道不是主反应通道。
2.2.3 抽提NH反应
反应物中间体IM1生成时,能量降低0.1kJ·mol-1,在IM1中,通过Si、S、C相互作用经TS1形成稳定的三元环中间体IM2,反应势垒为113.8kJ·mol-1。然后IM2经TS2生成产物P1(SCSiFCNH),反应势垒为113.2kJ·mol-1。在整个反应过程中C-N键长从0.1194nm(IM1)逐渐伸长至0.1295nm(TS2)、0.1382nm(IM2),同时,N-Si键长逐渐缩短,依次从0.3641nm(IM1),0.1901nm(TS1),0.1843nm(IM2),0.1609nm(P2),C-N的削弱有利于C-N键的断裂和C-Si键的生成。由表1所示,N-Si键BCP的ρ(r)逐渐升高至0.1607(P1),表明键强度逐渐增强,直至键生成。▽2ρ(r)值逐渐增大,表明N-Si键离子性增强。C-N键BCP的ρ(r)从0.4315(IM1)降至0.3024(IM2),表明键强度逐渐减弱,直至断裂生成P1。C-N键的▽2ρ(r)值逐渐增加,表明键的共价性逐渐减弱,离子性逐渐增强,形象地描述了键的断裂最后生成P1的过程。
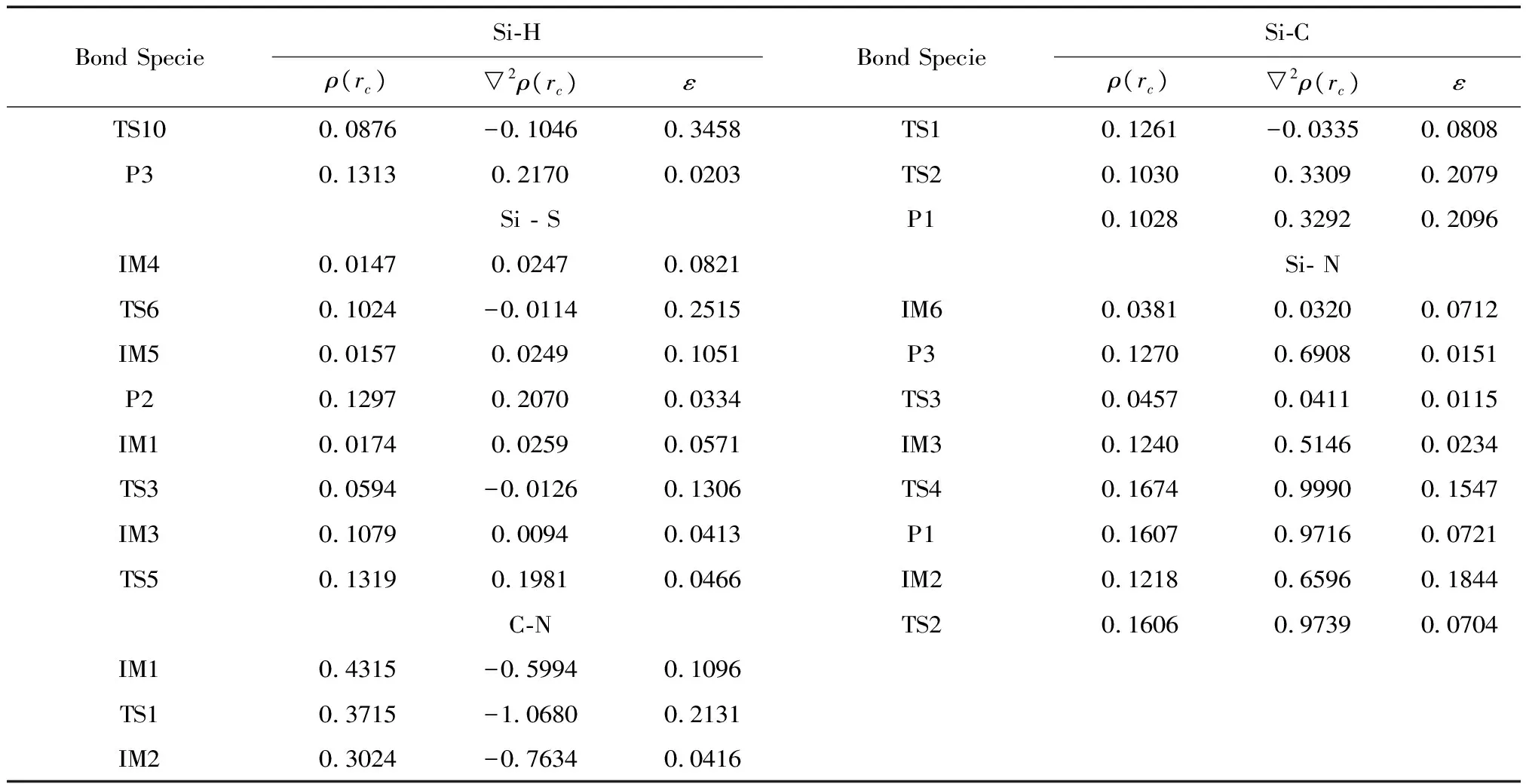
表1 反应通道主要化学键的电子密度拓扑性质
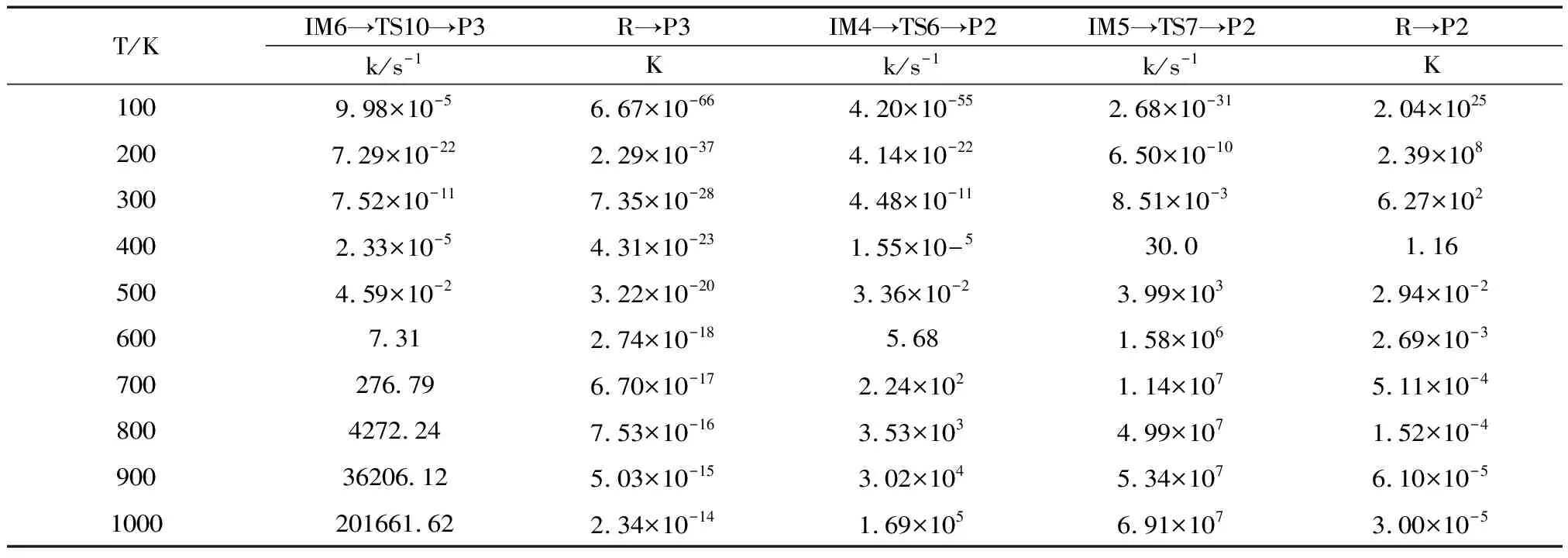
表2 1.0Atm下,在100-1000K内反应的平衡常数K及反应速率常数k(s-1)值
IM1经过渡态TS3形成稳定的四元环中间体IM3,反应势垒为91.7kJ·mol-1,IM3经TS4生成P1。整个反应吸热43.0kJ·mol-1。Si-N键长从0.4408nm(IM1)逐渐缩短至0.1609nm(P1),但C-N键长从0.1191nm(IM1)逐渐拉伸至0.2370nm(TS4),这非常有利于Si-N键生成,C-N键断裂。IM1经TS8生成三元环中间体IM2,IM2经TS9形成四元环中间体IM3,反应势垒较高,分别为158.2kJ·mol-1和286.7kJ·mol-1,且均为吸热反应。与抽提S反应相比,无论在热力学上还是在动力学上都是不利的。所以通过三元环和四元环抽提NH反应都不是主反应通道。
3 结论
基于密度泛函理论(DFT),在B3LYP/6-311++G**水平上对SiFCl自由基与异硫氰酸(HNCS)的反应机理进行系统的理论研究,结果表明,HNCS与单重态的SiFCl自由基反应有八条可能的反应通道,分别为抽提S、抽提NH及H迁移反应。其中经过渡态TS7抽提S反应生成产物P2(SSiFClCNH)的反应,由于在100-600K温度区间内,此反应的平衡常数和速率常数均较大,且反应放热95.1kJ·mol-1,因此,该反应为单重态主反应通道。本研究可能为除去光化学反应产生的卤代烃及其中间体等一些空气污染物的反应提供理论指导。
