奇亚籽油微胶囊的制备及表征
2020-03-28常馨月陈程莉董全
常馨月,陈程莉,董全
(西南大学 食品科学学院,重庆,400715)
奇亚籽(Chia seed)为芡欧鼠尾草的种子,属唇形花科,又名山香籽、鼠尾巴草种子,原产于墨西哥中部、南部和危地马拉等地区[1],目前主要种植于墨西哥、玻利维亚、厄瓜多尔和危地马拉等地[2]。奇亚籽油作为一种新型功能性油脂,种子含油量约为25%~38%[3],其α-亚麻酸(ALA)、亚油酸(LA)含量高达80%以上,是人体补充ω-3脂肪酸的油脂来源[4-5]。现代研究表明,富含多不饱和脂肪酸的饮食可以降低许多疾病的风险,尤其是心血管疾病、癌症和代谢综合征,因此日益受到人们的重视[6-7]。此外,奇亚籽油中还含有甾醇、生育酚、角鲨烯、绿原酸、槲皮素等多种活性物质,高含量的ALA和抗氧化物质, 使奇亚籽油具有调节血脂、抗氧化、促进肿瘤细胞凋亡等多种生理活性[8],对于预防冠心病、炎症、哮喘、视网膜等疾病和维持脑功具有重要的意义[9-10],因此,奇亚籽油具有较高的营养价值, 是一种具有良好潜力的保健品资源,具有很大的开发和应用前景。从营养角度看,奇亚籽油中富含多不饱和脂肪酸,对人体健康有积极的影响;然而,当这些产品与氧、光、湿、热接触时,就会发生脂质氧化,产生挥发性化合物,从而改变它们的化学结构和感官质量,导致其货架期缩短[11-12]。因此,延缓奇亚籽油的氧化对其功效和食用安全具有重要意义。
微胶囊技术不仅能有效防止奇亚籽油在储存和加工的不良反应,减少外部环境对奇亚籽油的氧化作用,还能有效控制油脂在存放期间风味的释放,改善消化吸收率,延长产品货架期[13]。现阶段微胶囊化的方法主要有:复合凝聚技术[14]、喷雾技术(喷雾干燥法、喷雾冷却法和电喷雾法)[15]、共挤出技术[16]、真空冷冻干燥技术[17]等。其中,冷冻干燥技术是在真空环境中将芯材冻结在共结晶点以下,使其凝固,并在极低压力下提供热量使物料中的水升华,从而使物料脱水的方法[18]。为了保证奇亚籽油的综合利用价值,本试验探究了奇亚籽油微胶囊的制备工艺参数,并对获得的微胶囊产品的理化性质进行研究,旨在为奇亚籽油产品的开发提供依据。
1 材料与方法
1.1 材料与试剂
奇亚籽油,西安明朗生物技术有限公司;酪蛋白酸钠、D-乳糖水合物(食品级),山东西亚化学工业有限公司;石油醚、无水乙醇(分析纯),成都市科隆化学品有限公司;无水乙醚(分析纯),扬州三和化工有限公司。
1.2 仪器与设备
冷冻干燥机,北京松源华兴科技发展有限公司;胶体磨,温州昊星机械设备制造有限公司;集热式恒温加热磁力搅拌器,巩义市予华仪器有限责任公司;旋转蒸发仪,上海亚荣生化仪器厂;电热鼓风干燥箱,天津赛得利斯试验分析仪器制造厂;箱式电阻炉,上海一恒科学仪器有限公司;凯氏定氮仪,上海沛欧分析仪器有限公司;医用离心机,长沙高新技术产业开发区湘仪离心机仪器有限公司;马尔文激光粒度分析仪,英国马尔文仪器公司;Phenom Pro扫描电镜,Phenom World公司;Spectrun 100傅里叶红外光谱仪,美国PerkinElmer公司;DSC 4 000差示扫描量热仪,美国PerkinElmer公司。
1.3 方法
1.3.1 奇亚籽油微胶囊的工艺流程
酪蛋白酸钠+D-乳糖-水合物→溶解→调配→奇亚籽油→恒温磁力搅拌均匀→胶体磨乳化→冷冻干燥→奇亚籽油微胶囊
1.3.2 单因素试验
选择壁材比[m(酪蛋白酸钠)∶m(D-乳糖-水合物)]、乳化温度、固形物浓度、乳化时间和壁芯比(质量比)作为评价包埋率的主要因素,采用单因素试验,探究其对奇亚籽油微胶囊效果的影响。
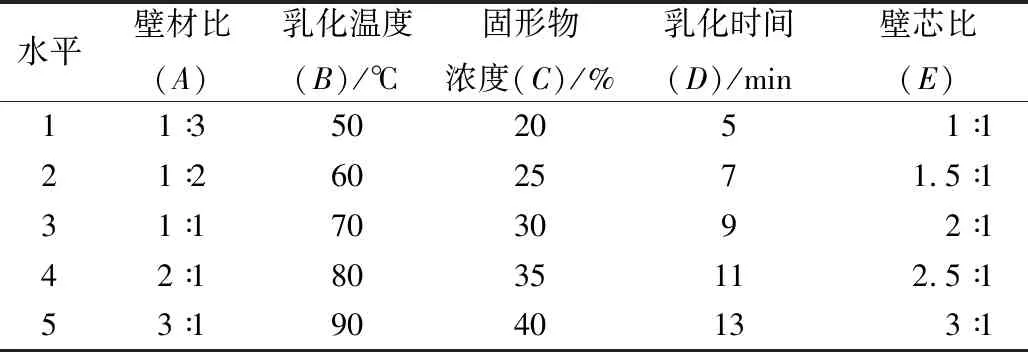
表1 单因素实验因素及水平表
1.3.3 奇亚籽油微胶囊包埋率的测定
1.3.3.1 微胶囊表面油含量的测定
1.3.3.2 微胶囊总油含量的测定
参考李成忠等[19]的方法并有所改进。称取1.0 g(准确至0.000 1 g)奇亚籽油微胶囊产品放入洁净干燥的烧杯中,加入热水20.0 mL,充分搅拌溶解后,依次加入无水乙醇、石油醚、无水乙醚(体积比2∶1∶1),搅拌静置后,将上层萃取液移入干燥至质量恒定的圆底烧瓶内,重复萃取2次,将萃取液归并入圆底烧瓶,在50 ℃条件下旋转蒸发除去溶剂,放入烘箱中烘至恒重,称量并计算得到总油含量。
1.3.3.3 包埋率的计算[20]

(1)
式中:m1,微胶囊产品表面油含量;m2,微胶囊产品总油含量。
1.3.4 理化性质
1.3.4.1 水分测定
称取1~2 g微胶囊产品置于烘箱中,在105 ℃下烘干至恒重,水分含量为烘干前后质量减少的百分数。
1.3.4.2 灰分测定
采用550 ℃灼烧法。参考《GB/T 5505—2008粮油检验 灰分测定法》。
1.3.4.3 粗蛋白测定
参考GB5009.5—2010。
1.3.4.4 流动性测定
准确称取10 g奇亚籽油微胶囊样品倒入漏斗,使微胶囊通过漏斗自然下落,在水平圆板上堆积,进行3次平行试验。测量粉堆高度H及粉堆覆盖半径R,按公式(2)求出休止角,休止角越大散落性越好,休止角越小散落性越差。
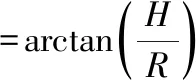
(2)
1.3.4.5 堆密度的测定
准确称取3 g奇亚籽油微胶囊样品,缓慢地装入有刻度的量筒中,并将量筒水平匀速晃动使微胶囊粉末自然下沉,测定体积,并计算单位体积微胶囊的质量即微胶囊的堆积密度。每一样品进行3次平行试验。微胶囊堆密度按公式(3)计算:
(3)
式中:ρ,微胶囊堆密度,g/cm3;M,微胶囊质量,g;V,容器容积,cm3。
1.3.4.6 溶解度的测定
已知微胶囊的水分含量m,准确称取一定质量(W)的微胶囊产品放入50 mL的离心管中,加入20 mL蒸馏水,在漩涡振荡仪上振荡2 min,使微胶囊充分溶解,然后在4 500 r/min离心15 min,除去上层溶液,再用少量的水把沉淀转移到已知质量的称量皿 (W1)中,在105 ℃烘箱中干燥至恒重(W2)。溶解度按公式(4)计算:

(4)
1.3.5 粒径分布
将待测微胶囊样品用蒸馏水稀释至一定浓度,用激光粒度分析仪进行粒度分析,得到微胶囊的粒径分布,并绘制颗粒大小分布曲线图。
1.3.6 表面结构观察
在样品台上贴上一层导电胶带,将微胶囊粉末均匀平铺在导电胶带上,样品经喷金处理后用扫描电子显微镜观察,加速电压为10 kV,放大倍数分别为5 000、10 000倍。
1.3.7 红外光谱分析
分别取2 mg的酪蛋白酸钠,D-乳糖水合物,奇亚籽油,冷冻干燥后的微胶囊样品,加入200 mg的溴化钾(105 ℃烘干)研磨压片,放于红外仪上进行扫描分析。扫描范围为400~4 000 cm-1,光谱分辨率2 cm-1,扫描次数64次。
1.3.8 DSC测定
用镊子分别称取2~4 mg的酪蛋白酸钠,D-乳糖水合物,奇亚籽油,微胶囊样品放入坩埚中,压片后用差式扫描量热仪测定。测定条件为:扫描温度范围20~250 ℃,升温速率为10 ℃/min。
1.4 数据统计与分析
利用Oigin软件(Vertion 9.0)对单因素试验折线图进行分析,利用Design-Expert(Vertion 8.0.6)对响应面试验进行线性回归和方差分析(P<0.05)。
2 结果与分析
2.1 单因素试验结果与分析
2.1.1 壁材比的影响
由图1可以看出,随着壁材比的增大,微胶囊包埋率先迅速增大,后逐渐减小。当壁材比为1∶1时,包埋率最高。当壁材比增加到1∶1以上时,微胶囊包埋率随着壁材比的增加而快速降低,这是因为一方面酪蛋白酸钠是一种表面活性剂,提高一定的浓度,可以降低油-水界面的张力,容易形成平衡、稳定的乳状液。但是酪蛋白酸钠也具有凝胶性,过量后会导致乳化液黏度增加,阻碍芯材和壁材的结合,影响均质效果,进而影响包埋率。另一方面随着壁材比的增加,D-乳糖-水合物含量降低,不能有效地包埋奇亚籽油与酪蛋白酸钠形成的络合物,使包埋率降低。因此选择壁材比1∶2~2∶1进行优化试验。

图1 壁材比对奇亚籽油微胶囊包埋率的影响
2.1.2 乳化温度的影响
从图2可知,随着乳化温度的升高,微胶囊包埋率先逐渐增大后逐渐减小,在乳化温度为70 ℃,微胶囊的包埋率达到最高。可能是因为温度过低不利于酪蛋白酸钠和D-乳糖-水合物的充分溶解,使得乳化效果差,导致包埋率低;当温度过高时,壁材的溶解度较温度低时高,但温度过高会造成蛋白质变性,不利于乳化,导致包埋效果不好。因此选择乳化温度70 ℃进行优化和验证试验。
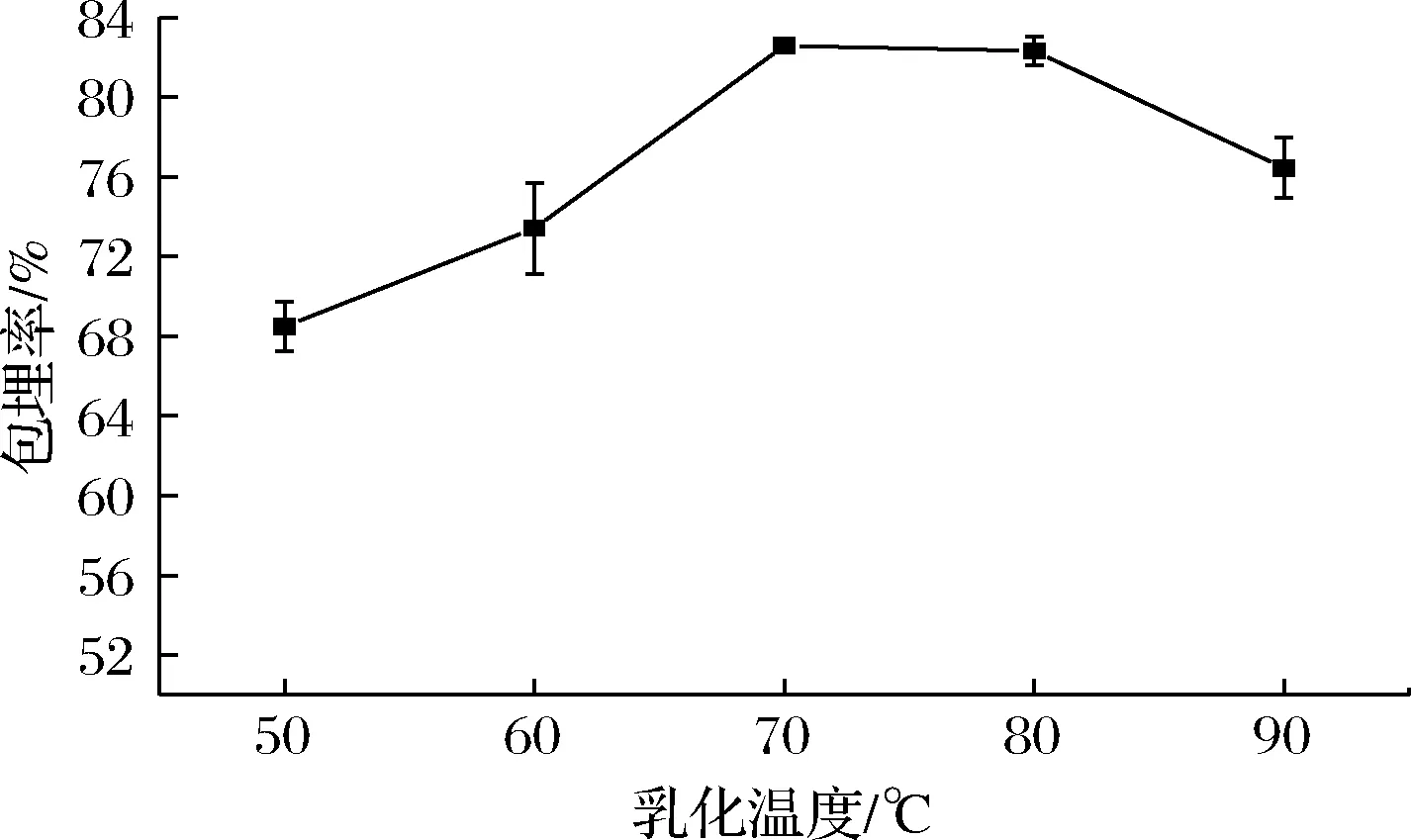
图2 乳化温度对奇亚籽油微胶囊包埋率的影响
2.1.3 固形物浓度的影响
如图3所示,固形物浓度对微胶囊化的影响是显著的。随着固形物浓度的增加,微胶囊包埋率迅速增加;当浓度超过一定范围时,随着固形物浓度的增加,微胶囊包埋率急剧下降。当固形物浓度达到30%时,包埋率最高。这可能是因为当固形物浓度逐渐增加时,溶液中的芯材和壁材的含量均增加,形成的微胶囊具有较好的致密性和强度。但当固形物浓度过高时,容易使乳化液黏度过高,不利于均匀化,均质效果差,导致包埋率降低。因此选择固形物浓度 25%~35%进行优化试验。

图3 固形物浓度对奇亚籽油微胶囊包埋率的影响
2.1.4 乳化时间的影响
如图4所示,在一定范围内,随着乳化时间的延长,微胶囊包埋率没有明显的增减趋势,总体水平保持在82%~88%。乳化时间为9 min时包埋率达到最大。这可能是因为随着乳化时间的延长,温度逐渐升高,乳化液黏度下降,微粒子变动,使得微胶囊稳定性和微粒子稳定性受到极大影响,从而导致微胶囊包埋率下降。因此选择乳化时间9 min进行优化和验证试验。

图4 乳化时间对奇亚籽油微胶囊包埋率的影响
2.1.5 壁芯比的影响
如图5所示,包埋率随壁芯比的增大而逐渐增大,在2∶1时达到最大值,当超过2∶1时包埋率迅速下降。这可能是因为随着壁芯比的增加,壁材含量升高,可以达到较好的包埋效果,包埋率也随之升高;但当壁芯比过大,壁材含量过多,不能完全包埋住芯材,形成的乳化液稳定性不佳,黏度增大,容易造成壁材的浪费,导致包埋率降低。因此选择壁芯比1.5∶1~2.5∶1进行优化试验。
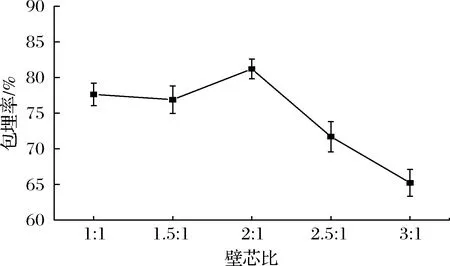
图5 壁芯比对奇亚籽油微胶囊包埋率的影响
2.2 响应面试验结果与分析
根据单因素试验结果,综合考虑各因素对奇亚籽油微胶囊包埋率的影响,各因素的选择水平见表2。

表2 响应面试验因素水平表
利用Design-Expert分析软件,采用B0x-Behnken设计试验,以壁材比、固形物浓度、壁芯比为自变量,包埋率为响应值,对奇亚籽油微胶囊的制备条件进行优化。试验结果见表3。从表3可知,包埋率最高的1组为试验6。
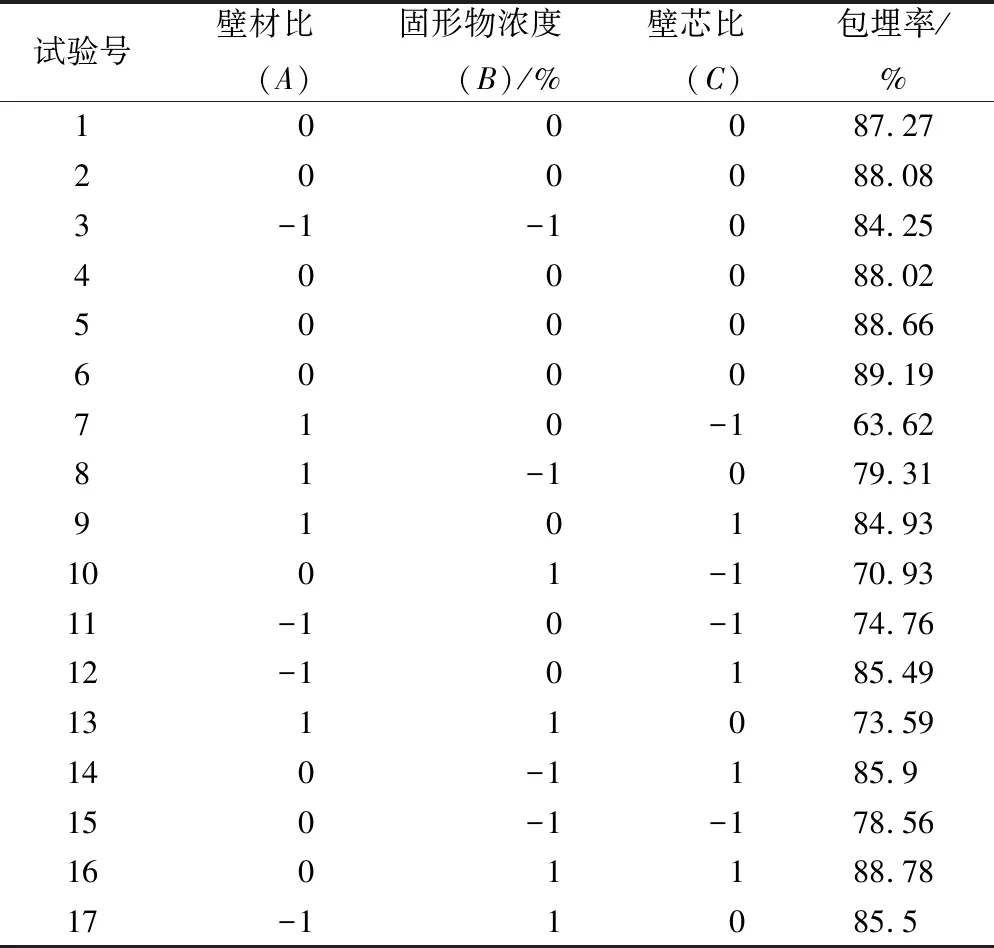
表3 响应面实验方案及结果
2.2.1 微胶囊包埋率的回归方程及方差分析
软件分析结果得到二次多项式回归方程为:
Y=88.24-3.57A-1.15B+7.15C-1.74AB+2.65AC+2.63BC-5.71A2-1.87B2-5.33C2
由表4可知,以微胶囊包埋率为响应值时,该模型极显著(P<0.000 1),失拟项不显著(P=0.060 4>0.05),说明模型预测值与实际误差较小,即该回归模型能够较好地拟合壁材比、固形物浓度及壁芯比对奇亚籽油微胶囊包埋率的影响情况。回归方程中所述各因素与响应值之间存在显著关系(R2=0.987 4),表明微胶囊包埋率的变化有98.74%来源于所选变量,说明该模型与实际拟合较好。
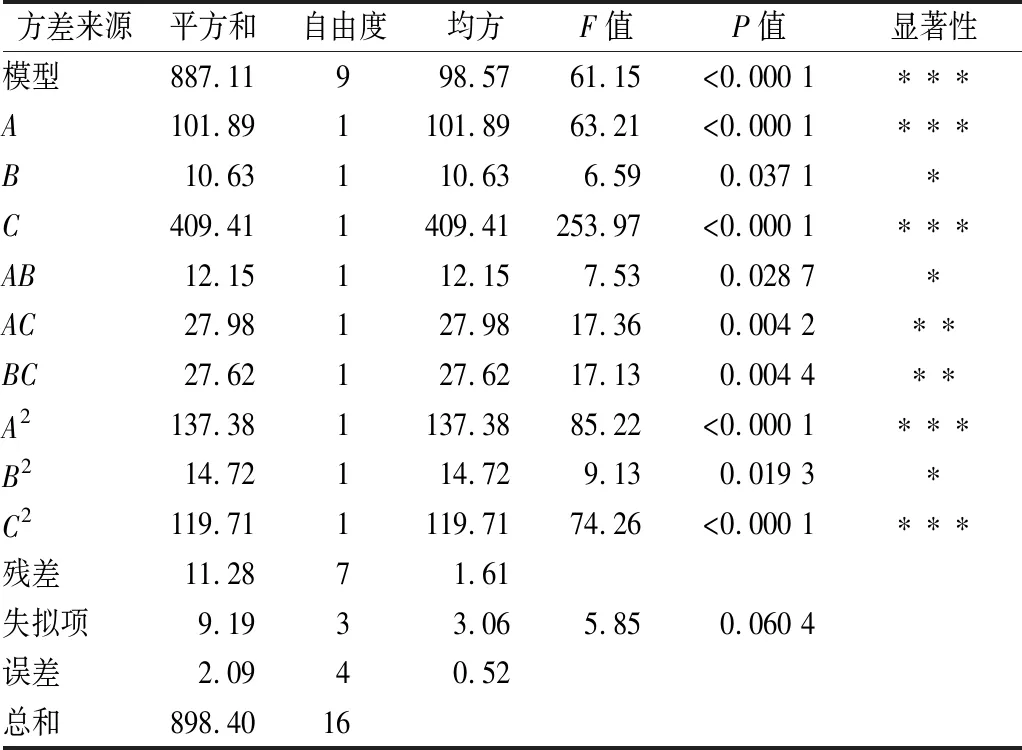
表4 回归模型的方差分析
注:***表示极显著性差异(P<0.000 1);**表示高度显著性差异(P<0.01);*表示显著性差异(P<0.05)
方差分析结果表明:A、C、A2、C2呈极显著影响(P<0.000 1),AC、BC呈高度显著影响(P<0.01),B、AB、B2呈显著影响(P<0.05)。说明壁材比例、固形物浓度和壁芯比均对奇亚籽油微胶囊的包埋率有一定的影响,就影响程度上,壁芯比>壁材比>固形物浓度。
2.2.2 响应面结果分析
通过分析壁材比A、固形物浓度B和壁芯比C3个因素的交互作用对奇亚籽油微胶囊包埋率的影响,分别得到各因素交互作用关系的响应面图和等高线图,参见图6、图7、图8。
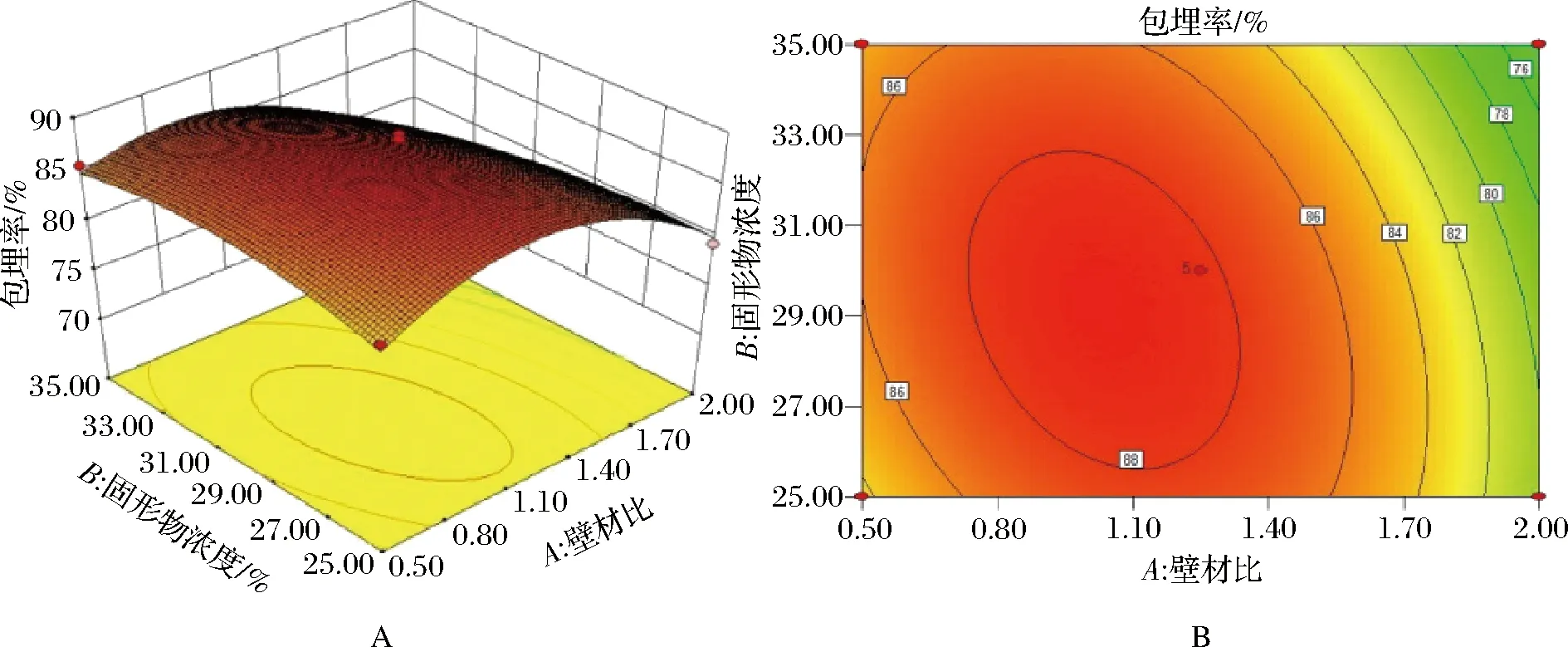
图6 壁材比及固形物浓度对包埋率的影响
由图6可知,在一定范围内,当固形物浓度一定时,包埋率随壁材比的增大先升高后降低;当壁材比一定时,包埋率随固形物浓度的增大先升高后减小,说明壁材比和固形物浓度均对包埋率有影响。当壁材比或固形物浓度确定时,固形物浓度的坡度更为平缓,说明壁材比对奇亚籽油包埋率的影响大于固形物浓度。等高线由高水平到低水平逐渐密集,与其他2个图相比,图6中的等高线更趋于圆形,坡度最为平缓,说明壁材比与固形物浓度之间的交互作用对包埋率的影响最小。
从图7可以看出,微胶囊包埋率随壁材比和壁芯比的变化呈现比较陡峭的坡度,说明壁材比和壁芯比均对包埋率影响较大。当壁材比或壁芯比确定时,壁材比的坡度较为平缓,说明壁芯比对奇亚籽油包埋率的影响大于壁材比。图中等高线的分布趋于椭圆,说明壁材比与壁芯比的交互作用对包埋率呈高度显著的影响。
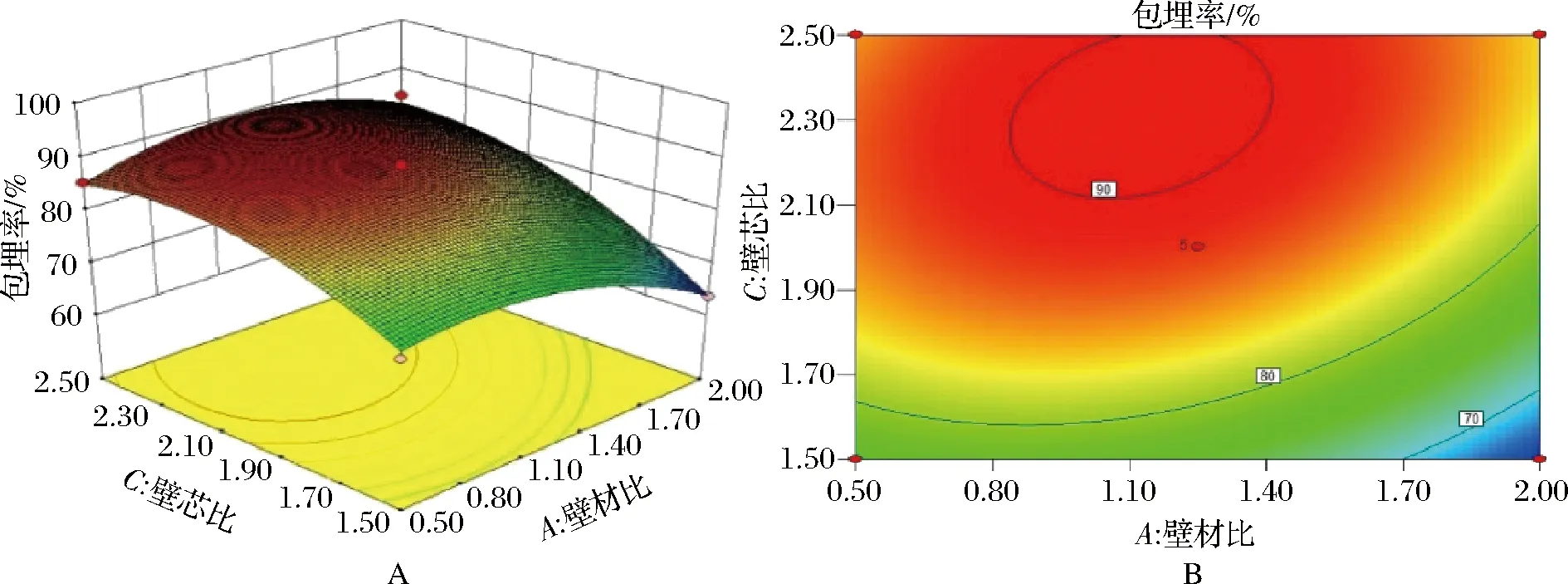
图7 壁材比及芯壁比对包埋率的影响
由图8可以看出,固形物浓度和壁芯比的变化也会使微胶囊包埋率呈现比较陡峭的坡度,说明固形物浓度和壁芯比对包埋率均有较大的影响。当固形物浓度或壁芯比确定时,固形物浓度的坡度更为平缓,说明壁芯比对奇亚籽油包埋率的影响大于固形物浓度。图中等高线呈椭圆状,说明固形物浓度与壁芯比的交互作用对包埋率有显著影响。在本实验中,不同因素的交互作用对奇亚籽油微胶囊包埋率的影响为:AC>BC>AB。因此,在冷冻干燥法制备奇亚籽油微胶囊的实际生产过程中,在乳化温度和乳化时间固定的条件下,通过控制壁材比、固形物浓度、壁芯比等因素,可以提高包埋率。

图8 固形物浓度及壁芯比对包埋率的影响
2.3 验证试验
通过Design-Expert.V 8.0.6软件得到最佳工艺参数为壁材比1.1∶1、固形物浓度31.32%、壁芯比2.34∶1,在此工艺条件下,微胶囊的理论包埋率为90.90%。在最佳条件下,进行3次平行实验,得到平均包埋率和预测精度分别为90.65%和99.72%,说明预测值与实验值接近,表明该模型是有效的。
2.4 奇亚籽油微胶囊的理化性质
奇亚籽油微胶囊产品呈疏松粉末状,色泽呈乳白色,无杂质颗粒或酸败等异味。从表5可知,奇亚籽油微胶囊中水分含量为(4.46±0.01)%,表明微胶囊产品水分含量低,不易黏结成块发生霉变。根据休止角和流动性的对应关系,30°~45°即产品的流动性较好,由于奇亚籽油微胶囊的休止角为(36.71±0.11)°,堆密度为(0.21±0.01) g/cm3,说明制备得到的微胶囊的流动性较好,粉末表面光滑,黏度小。微胶囊表面油含量为(2.82±0.15)g/100 g,总油含量(30.16±0.11)g/100 g,微胶囊产品表面含油量少,表明在包埋过程中奇亚籽油挥发少,微胶囊表面甚少破损,奇亚籽油大都被酪蛋白酸钠和D-乳糖-水合物给包埋住了,从而保证了微胶囊的总油含量和质量。

表5 奇亚籽油微胶囊的理化性质
2.5 奇亚籽油微胶囊粒径分布分析
由图9可知,奇亚籽油微胶囊的粒径主要分散在0.1~1 μm范围内,符合一般生产对微胶囊粒度的要求,微胶囊的平均粒径在426.4 nm 左右,多分散系数(PDI)值为0.468,较小的PDI值证明奇亚籽油微胶囊粒径大小均匀,分布较为集中。

图9 奇亚籽油微胶囊的粒径分布
2.6 微胶囊表面结构分析
对最佳制备工艺条件下得到的微胶囊化粉体进行了扫描电镜观察,微胶囊外观形貌见图10。冷冻干燥过程被认为是一种生产高质量干燥食品的方法,因为干燥过程是在真空和低于环境温度下进行的。这一特性使得冷冻干燥对热敏性和生物活性成分如奇亚籽油的干燥特别有吸引力,最大限度地减少了喷雾干燥过程中高温对产品的损害。由图10-a可知,以酪蛋白酸钠和D-乳糖-水合物为壁材制备的奇亚籽油微胶囊呈现不规则的几何形状和紧凑的结构,不同于喷雾干燥制备得到的球形、规则的微胶囊形状。从图10-b中可以看到,在微胶囊表壁上存在一些突起和小气孔,这可能是因为,在冻结过程中自由水形成冰晶,冰晶在冻结过程中产生了空洞。

a-微胶囊的不规则形状;b-微胶囊的表壁结构
2.7 微胶囊红外光谱分析

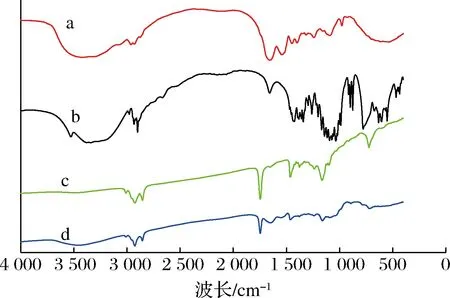
a-酪蛋白酸钠;b-D-乳糖-水合物;c-奇亚籽油;d-奇亚籽油微胶囊
2.8 微胶囊DSC分析
由图12可知,D-乳糖-水合物和酪蛋白酸钠发生相转变的起始温度分别为144.17、106.59 ℃;吸热峰分别为150.31、126.82 ℃。

a-D-乳糖-水合物;b-酪蛋白酸钠;c-奇亚籽油;d-奇亚籽油微胶囊
由图12d曲线可知,奇亚籽油微胶囊发生相转变的起始温度为121.77 ℃,在该温度前,微胶囊处于玻璃态,性质较稳定,结构还未发生变化。可以看出微胶囊的玻璃化转变温度较高,表明此物质较稳定。随温度的继续升高,微胶囊产品中的酪蛋白酸钠和D-乳糖-水合物在高温下受热溶胀,各反应速率也相应的增加,此时有序的晶体结构向无序的晶体结构转变,奇亚籽油微胶囊发生热熔解,其峰值为131.17 ℃。由于奇亚籽油微胶囊的热熔解温度较高,因此,在常规的热加工处理过程中,奇亚籽油微胶囊的结构仍然完整。
3 结论
(1)以酪蛋白酸钠与D-乳糖-水合物为壁材,以奇亚籽油为芯材,采用冷冻干燥法制备奇亚籽油微胶囊。在单因素试验的基础上,以微胶囊的包埋率为响应值,以壁材比、固形物浓度和壁芯比为因素值,建立了二次回归模型方程。3个因素对奇亚籽油微胶囊包埋率的影响顺序为:壁芯比>壁材比>固形物浓度。采用响应面法优化微胶囊化工艺,最佳工艺参数为:壁材比(酪蛋白酸钠∶D-乳糖-水合物)1.1∶1(质量比)、固形物浓度31.32%、壁芯比2.34∶1,在此条件下,奇亚籽油微胶囊包埋率达90.65%。(2)通过测定微胶囊产品的基本理化性质,表明制备得到的奇亚籽油微胶囊水分少,流动性较好,粉末表面光滑,黏度小。(3)微胶囊产品粒径大小均匀,主要分散在0.1~1 μm范围内。(4)微胶囊呈不规则的几何形状和紧凑的结构,表壁上存在一些突起和小气孔。(5)芯材、壁材的主要特征吸收峰在微胶囊产品的红外光谱图上均有所显示,只是强度有所减弱,表明奇亚籽油微胶囊的包埋结构形成。(6)经DSC分析表明,当温度达到131.17 ℃时微胶囊发生热分解,热稳定性良好,基本可以满足一般食品加工条件。
