基于立方烷结构的分子催化剂在光催化水氧化中的研究进展
2020-03-27孙万军林军奇梁向明杨峻懿马宝春丁勇
孙万军,林军奇,梁向明,杨峻懿,马宝春,丁勇,2,*
1 兰州大学化学化工学院,兰州 730000
2 中国科学院兰州化学物理研究所,羰基合成与选择氧化国家重点实验室,兰州 730000
3 甘肃民族师范学院化学与生命科学系,甘肃 合作 747000

1 引言
当今世界,随着世界人口的不断增加以及工业化的飞速发展,人们对能源的需求也越来越大。加之传统化石燃料(煤、石油、天然气)的不可再生性和大量使用过程中所引起的环境污染等问题日益严重,发展可再生能源和提高能源利用率迫在眉睫1,2。太阳能作为一种绿色、可持续的清洁能源受到全世界的广泛关注3-5。利用太阳能进行光催化水分解是解决该问题的有效途径之一,然而水的两个半反应有着截然不同的机理,包括质子还原和水的氧化式(1-3),其中水的氧化是一个需要高能量逐步反应的四电子转移过程(237 kJ·mol-1)以及O―O键的形成显得极其复杂,因此,水的氧化反应是自然光合作用和人工光合成的速控步骤6-8。

开发高效稳定的水氧化催化剂是实现该过程的 关 键9。 自1982 年, Meyer 报 道 的cis,cis-[(bpy)2(H2O)Ru(m-O)Ru(H2O)(bpy)2]4+(bpy = 2,2’-联吡啶)首个以双钌为水氧化分子催化剂以来10,为水的分解开启了先河。在自然界光合作用中,光系统PSII过程也就是水氧化反应过程。科学家们已经证明PSII中水氧化反应的活性位点是Mn4CaO5,该结构由三个Mn和一个Ca原子组成的变形四面体Mn3CaO4,第四个Mn则是通过氧桥键与该四面体相连11。2015年,张纯喜等12首次成功地合成获得了结构与自然界光合裂解水催化中心Mn4Ca簇类似的人工模拟物[Mn4CaO4(ButCO2)8(ButCO2H)2(py)] (But= tert-butyl,py = pyridine)。该化合物是迄今为止所有人工模拟物中与生物水裂解催化中心结构最为接近的模拟物(图1),它不仅很好模拟了不对称的Mn4Ca簇核心结构,而且模拟了其生物配体环境。该模拟物中四个Mn离子的价态(+3,+3,+4,+4)与生物水裂解催化中心完全一致,其氧化-还原特性、电子顺磁特性及化学反应特性方面也均与生物水裂解催化剂类似,而且同样具有催化水裂解的催化功能,开启了人工光合作用水裂解研究的新篇章。同时OEC催化产氧机理也被广泛研究。结果表明,该催化循环过程包含五个氧化态,即S0-S4态(Kok循环)13。S0为最还原的状态,也就是催化循环的静息状态。S0态的Mn4CaO5簇会逐步失去电子被氧化至S4态,即最高氧化态,S4态还原到S0态的还原过程中,完成O2的释放(图2)14,15。
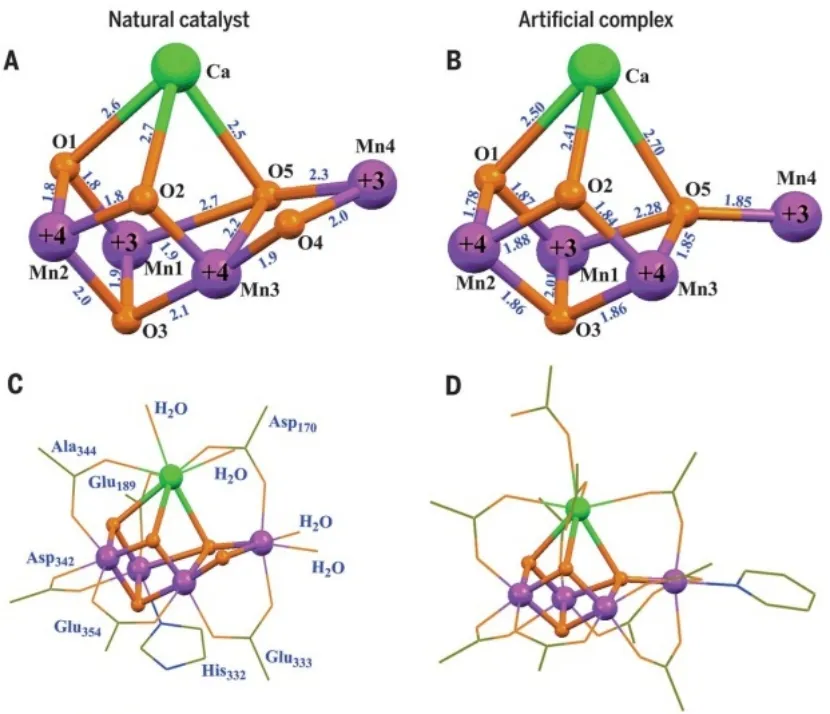
图1 OEC 中Mn4CaO5 簇(A,C)和合成的立方烷结构(B,D) 12Fig. 1 Structures of the native OEC (A, C) and the synthetic Mn4Ca complex (B, D) 12.

图2 自然光合作用中的 Kok 循环示意图13Fig. 2 The Kok cycle in natural photosynthesis 13.
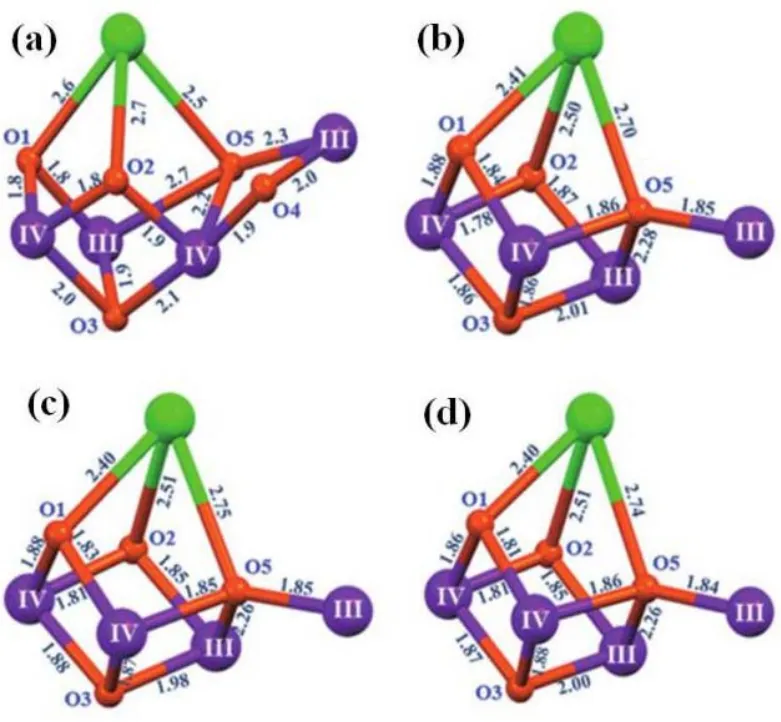
图3 OEC 中Mn4CaO4 簇(a)和合成的三种Mn4CaO4 簇(b-d) 16Fig. 3 Structures of the OEC (a) and three artificial Mn4CaO4 complexes (b-d) 16.
最近,张纯喜课题组16成功制备出能够在极性溶剂中稳定存在的新型仿生Mn4CaO4簇合物,实现对光合作用水裂解催化中心更精确模拟(图3)。该工作成功模拟了生物OEC的核心骨架和配体环境及其氢键相互作用,并在关键辅基Ca离子上引入可交换的溶剂分子(乙腈或N,N-二甲基甲酰胺),这类新型Mn4CaO4簇合物在极性有机溶剂中可稳定存在,为人工光合作用仿生水裂解催化剂的功能和应用研究提供了重要保障。此外,这类仿生Mn4CaO4簇合物的合成和结构解析也为理解自然界光合作用水裂解催化中心的结构和催化机理提供了重要线索。
受自然界光合作用的启发,通过模拟自然界光合作用,实现太阳能的有效转化,是解决人类面临的能源危机和环境问题的有效途径之一。为此,科学家一直致力于开发高效、稳定、廉价,含有立方烷结构的分子催化剂,由于其催化活性比较好,且类似于自然界中Mn4CaO4的仿生结构,受到了研究者的广泛关注17。
为了更加有效地实现产氧的进行,光催化水氧化反应中需要有牺牲电子受体的存在18,常见的牺牲电子受体有NaIO3和AgNO3等。光敏剂一般用[Ru(bpy)3]Cl2及其衍生物19,缓冲溶液一般有硼酸盐、磷酸盐等。本文中概述的体系主要为可见光/[Ru(bpy)3]2+/Na2S2O8水氧化体系,因此这里着重介绍这一体系的光催化循环过程。该体系的反应历程如图4所示。首先,光敏剂[Ru(bpy)3]2+被可见光激发,形成激发态的[Ru(bpy)3]2+*。随后Na2S2O8将[Ru(bpy)3]2+*氧化猝灭,原位生成氧化性很强的光生氧化剂[Ru(bpy)3]3+([Ru(bpy)3]3+/2+=接着[Ru(bpy)3]3+与水氧化催化剂反应,生成高价态的活性物种和H2O相互作用,经历一系列反应,最终释放出氧气。需要说明的是,的还原电势在2.4 V (vsNHE)以上21,一方面能直接将催化剂氧化,为保证硫酸根自由基阴离子优先和[Ru(bpy)3]2+发生反应,在该体系中光敏剂的浓度要远大于催化剂的浓度;另一方面,也能将[Ru(bpy)3]2+氧化为[Ru(bpy)3]3+,如式(4)和(5)所示。但式(4)所代表的反应过程较式(5)快很多。一般而言,我们用氧气收率,转化数(TON)、转化频率(TOF)以及量子效率(QE)几个参数来评价水氧化催化剂的标准。

图4 立方烷基光催化水氧化反应过程Fig. 4 The process of molecular cubanes catalyzed light-driven water oxidation.

综上所述,由于立方烷分子催化剂的结构、组成和性质具有可调性,因此探索具有明确结构的立方烷分子催化剂来实现从分子水平上模拟Mn4CaO5簇具有可行性。本文将按照基于有机配体的立方烷配合物和基于全无机的多金属氧酸盐立方烷水氧化催化剂分类进行讨论。
2 基于有机配体的立方烷配合物
2.1 钴为中心原子的配合物
钴基水氧化催化剂因其环境友好,来源丰富,热力学稳定、价格低廉以及催化效率高等优点被广泛关注19。
关于含有Co4O4立方烷结构的催化剂最早是由Dismukes课题组开展的。2011年,该课题组22报道了第一例完整的Co4O4立方烷分子催化剂Co4O4(OAc)4(py)4配合物1 (图5),该Co4O4核由四个双齿醋酸根配体和四个吡啶配体组成,其对称性属D2d点群。当光催化反应1 h时,TON为40,TOF为0.02 s-1,产生的氧气被认为是来自两个μ3-O桥的过氧键所释放出的,这与之前报道的含有Mn4O4立方烷结构的机理相似23。作者通过1H NMR检测光催化反应中该化合物的变化表明反应前后该立方烷的结构基本没有发生分解。

图5 配合物1 的球棍模型Fig. 5 The ball-and-stick representations of complex 1.
图6 立方烷配体与金属的结构排列示意图24Fig. 6 Structural permutations of ligand and metal nuclearity on the cubane core 24.
随后,该课题组24以钴簇配合物的配体以及金属中心两个方面研究了分子水氧化催化剂的活性(图6)。六种以吡啶或二吡啶配位的完整配合物1和离子型[Co4O4(OAc)2(bpy)4]2+(配合物1a)、半立方烷Co2O2二聚物{[Co2(OH)2(OAc)3(py)4](PF6)(配合物2)和[Co2(OH)2(OAc)3(bpy)2](ClO4)(配合物2a)}以 及 不 完 整 的 Co3O3, Co3O4三 聚 物{[Co3O(OH)2(OAc)3(py)5](PF6)2( 配 合 物 3) 和[Co3O(OH)3(OAc)2(bpy)3](ClO4)2(配合物3a)}的光催化产氧结果表明25,26,含有两个或三个金属簇的结构没有水氧化活性,完整的立方烷1和离子型1a的一级速率常数分别为0.030和0.015 s-1,说明不稳定羧酸根位点的数目越多越有利于氧气的释放。此外,Scandola等人27也对该化合物配合物1的光催化产氧活性进行了研究,其量子产率可达30%。
2012年,Bonchio等人28报道了一系列以吡啶衍生物为配体的立方烷结构的分子催化剂(μ-O)4(μ-CH3COO)4(p-NC5H4X)4](1,1b-1g,X = H,t-Bu,CN,Me,Br,COOMe和OMe,图7)。作者通过Hammett线性自由能曲线表明了配体给电子能力与光诱导电子转移常数之间的关系。这些配体影响了配合物的氧化还原性质,从而影响了光催化活性,量子效率从10% (X =t-Bu)增加到80%(X = OMe),表明吡啶配体上的给电子基团有利于与光生氧化剂的电子转移,从而增强了光催化产氧活性。
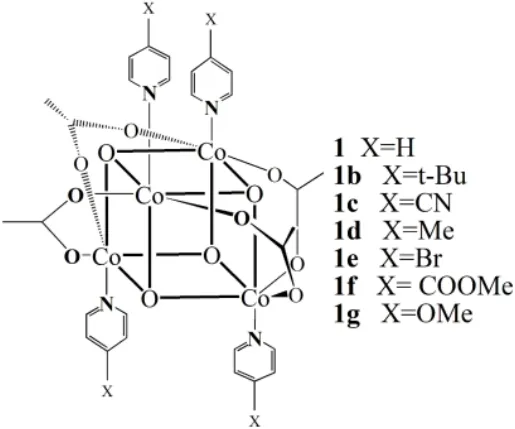
图7 配合物1,1b-1g 的结构示意图Fig. 7 Structural representation of complexes 1, 1b-1g.
2014年,李斐和孙立成课题组29将光敏剂[Ru(bpy)3]2+与含有立方烷结构的非贵金属分子催化剂配合物1通过羧酸根连接起来,分别合成了具有线性{Co4O4(OAc)3(Py)4}{(L)Ru(bpy)2}(L = bpy-4-CH3,4’-COOH) 和 环 形{Co4O4(OAc)2(Py)4}2{(L)Ru(bpy)2}2(L = bpy-4-COOH,4’-COOH))的两种构型的超分子自组装催化剂化合物4和化合物5(图8)。与分散体系相比,化合物4和化合物5的光催化产氧活性明显优于分散体系,且具有环状构型的催化剂的产氧活性是线性结构的5倍。同时稳定性测试表明,环状构型的催化剂的稳定性明显优于线性结构的稳定性。
Co4O4(OAc)4(py)4簇以及相关衍生物被广泛应用于光催化水氧化反应,然而2014年,Nocera课题组30通过电子顺磁共振光谱,核磁共振谱线增宽分析,微分电化学质谱法(DEMS)等手段发现主要的光催化产氧活性来源于在催化剂合成过程中引入少量的二价钴离子造成的,使用纯化后的催化剂只能检测到微量的氧气产生(图9)。Nocera等人的工作表明在研究Co配合物催化的水氧化反应时,必须排除在催化剂合成过程中引入杂质带来的影响。


图8 线性4 (a)和环形5 (b)的两种构型的钴基立方烷超分子自组装示意图Fig. 8 The structures of cobalt cubane with two supramolecular assemblies in linear 4 (a) and macrocyclic configurations 5 (b).
Tilley课题组31研究表明,尽管配合物1在较低pH和较低电势下是没有催化活性,但是在较强的碱性条件(pH > 10)以及较高的电势下能表现出较好的电催化水氧化活性,这意味着由于光催化过程中原位生成的氧化能力有限,需要更强的氧化条件将配合物1氧化才能完成该过程。同样具有光催化水氧化反应活性的双核钴核心结构的 多 吡 啶 配 合 物 [(TPA)CoIII(μ-OH)(μ-O2)CoIII(TPA)](ClO4)332被Thapper课题组报道。在随后的研究工作中,鲁统部课题组的工作33表明该配合物在电催化和光催化产氧过程中,会分解为CoOx起催化作用,配合物分子本身是没有水氧化活性的。这表明催化剂合成后对其纯化以及判断一个真正的分子催化剂是非常重要的。
鉴于以上工作,在研究立方烷配合物催化的光催化水氧化反应时,尤其是Co基配合物催化的水氧化反应时,一方面要保证催化剂的纯度,防止有Co2+杂质引入;另一方面,排除催化剂在光催化过程中配体分解导致金属离子游离出之后,变为活性物种。结合2015年Patzke课题组34以及2018年Finke课题组35的工作,我们可以设计一个思路来判断催化活性中心的来源(图10),为我们设计稳定高效的立方烷光催化剂提供新的思路。
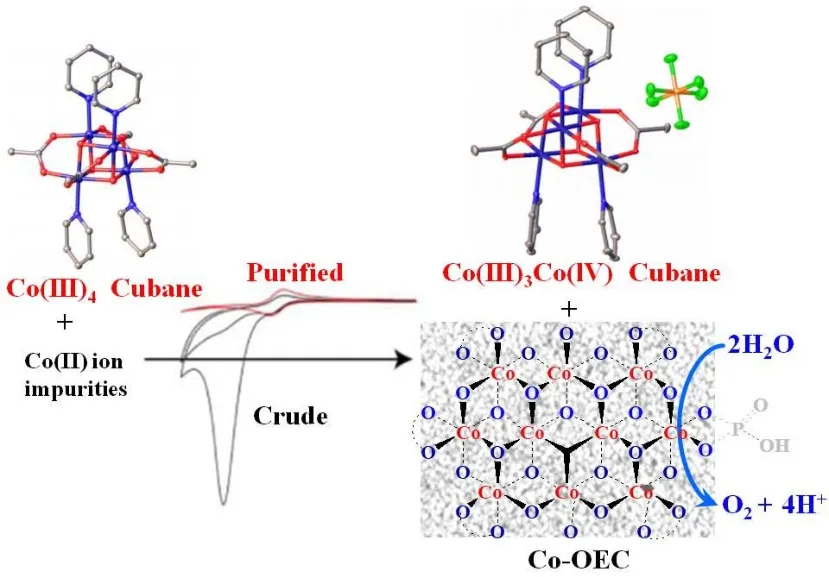
图9 水氧化活性来自1 中引入Co(II)离子30Fig. 9 The oxygen evolution of the 1 came from its Co(II) ion impurities 30.

图10 催化剂稳定性及活性中心的判断策略Fig. 10 Overall workflow and strategy for WOC stability tests.
2013年,Patzke课题组36报道了具有立方烷构型的2-(羟甲基)吡啶)作为分子水氧化催化剂(配合物6,图11)。该化合物首次将含二价钴Co(II)中心引入到一个立方烷结构的光催化水氧化催化剂中;立方烷核心的周围拥有柔性的配位水分子,通过脱质子化和配体交换提高了水氧化性能;两个醋酸配体通过单齿配位的形式连在Co(II)中心原子上;为模拟具有PSII产氧中心Mn4CaO4簇合物类似结构的水氧化催化剂的反应机理提供了实验的基础。在光催化实验中,TOF值随pH值的增加而增加,在pH 9.0时达到最高的TOF = 7.0 s-1,在pH 8.0时有最高的TON = 40。通过循环伏安、傅里叶变换红外光谱(FI-IR)、紫外可见吸收光谱(UV-Vis)以及动态光散射(DLS)等实验表明该催化剂在光催化反应后仍然保持着其完整的立方烷结构。
2015年,该课题组34在进一步的工作中将上述配合物6中的一个Co中心以镧系金属离子Ho、Er、Tm和Yb取代,得到了第一例以镧系元素Ln3+掺杂的Co基立方烷水氧化催化剂图12)。相比配合物6,配合物7a-7d 在可见光水氧化体系中的催化活性得到大幅度提升。该系列配合物结构中的Ln3+类似于PSII中Mn4CaO5簇中的Ca2+,其中配合物7b水氧化活性最好,催化TON和TOF分别为211和9 s-1(表1)。随后,作者通过一系列手段研究了中真正的催化活性物种。第一,稳定性测试以及排除纳米颗粒生成的测试,主要包括水氧化性能测试、Co2+沥出的验证、循环实验以及动态光散射实验;第二,痕量金属分析,包括乙二胺四乙酸对Co2+螯合以及结合电感耦合等离子体质谱分析;第三,催化剂的结构完整性测试,主要包括光谱测试、高效液相色谱分析、X射线近边吸收光谱/扩展X射线精细结构光谱(XANES/EXAFS)光谱分析以及高分辨质谱分析。上述多种测试手段表明催化剂在反应前后是稳定的。最后,基于Born-Oppenheimer近似计算和光催化实验结果表明,非常类似于Mn4CaO5簇中的Ca2+的作用机理。但是该催化剂7b在电催化反应完之后,扫描及能量色散X射线分析显示,其表面有一定的纳米颗粒生成,说明7b在电催化水氧化过程中发生了分解。这表明催化剂的稳定性一方面与分子结构本身有关,另一方面与具体反应条件有关,同一催化剂在不同的反应条件下会有不同的催化行为。

图11 配合物6 的球棍模型Fig. 11 The ball-and-stick representation of complex 6.
2017年,该课题37以二(2-吡啶基)甲酮(dpk)为配体,构筑了一系列结构新颖的具有Co2+和Ni2+金属中心立方烷结构的分子水氧化光催化剂Ni4-x(dpy{OH}O)4(OAc)2(H2O)2](ClO4)2(CoxNi4-x,其中配合物Co4O4记为8a,Ni4O4记为8b,图13),在最佳条件下,氧气收率最高可以达到80%。通过原位XANES表明:光催化水氧化经历了一个从CoII到CoIII或者到更高的氧化态,最后缓慢地回到初始的CoII。动态光散射实验表明在反应过程中没有纳米颗粒生成,同时基于分子动力学的密度泛函理论(DFT-MD)模拟计算和原位EXAFS测试表明,Co4O4在溶液中是稳定的,而其它CoxNi4-xO4-dpk的稳定性并未研究。随着镍含量的增加,光催化水氧化活性急剧下降,Ni4O4立方烷在相同的条件下光催化产氧活性很低。作者认为具有水配体的两个Co中心能充当水氧化活性位点,配位水能与分子结构中的羟基结构形成氢键,促进催化过程中O―O键的形成,在此过程中,Co中心价态会升高,至于是单一位点催化还是双金属中心协同催化,还有待进一步地研究。

图12 配合物7a-7d 的球棍模型Fig. 12 The ball-and-stick representation of complexes 7a-7d.

表1 各种立方烷结构的水氧化催化剂催化性能的总结Table 1 The summary of catalytic effects of various molecule cubanes WOCs.

图13 配合物8a 的球棍模型Fig. 13 The ball-and-stick representation of complex 8a.
2016年,孙頔课题组38报道了两例同构的分子中具有准立方烷结构片段的水氧化催化剂Co4L6X2(L = 1, 3-二甲基-5-羟甲基-1H-吡唑,X =Cl记为配合物9a,X = Br为配合物9b) (图14)。其对称性属单斜P21/n空间群,其中Co1和Co1i通过来自配体中吡唑上的两个氮原子和四个甲氧基中的氧原子配位形成八面体,而Co2和Co2i通过三个配体中的三个甲氧基上的氧原子,一个吡唑上的氮原子和一个卤素原子形成一个三角双锥体构型。在[Ru(bpy)3]Cl2和K2S2O8以及pH 8.0下,配合物9a和9b 的初始产氧速率分别为11.36 和2.18 mmol·s-1·g-1。
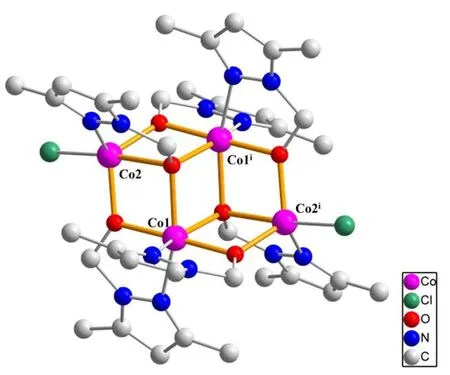
图14 配合物9a 的球棍模型Fig. 14 The ball-and-stick representation of complex 9a.
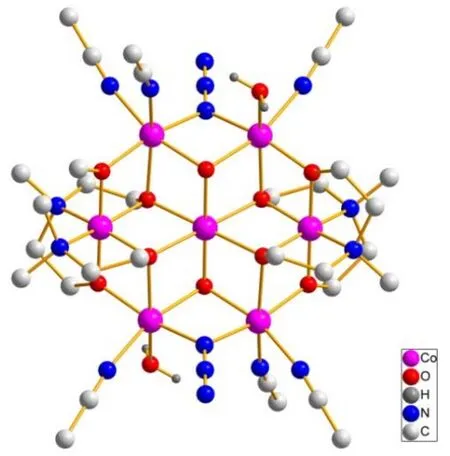
图15 配合物10 的球棍模型Fig. 15 The ball-and-stick representation of complex 10.
含有机配体的更高核配合物在光催化水氧化体系中的研究的比较少。随后该课题组39设计合成了一例具有碟状结构的七核混合价态Co基簇合物[Co5IICo2III(mdea)4(N3)2(CH3CN)6(OH)2(H2O)2]4-(H2mdea = N-甲基二乙醇胺)(配合物10,图15),其光催化水氧化的TON和TOF分别为210和0.23 s-1。发现加入螯合剂EDTA和2,2’-联吡啶对体系产氧活性的影响比较小,说明该七核簇合物中钴离子并不是主要的催化活性物种。进一步通过电喷雾电离质谱(ESI-MS)对该簇合物在溶液中及催化反应后的存在形式进行了细致地研究发现,其在水溶液中是以五核及四核钴的形式存在。当催化反应完成后,该簇合物中的有机配体发生解离,生成了核具有更高价态的这些钴物种可能是水氧化的催化活性物种。
2015年,我们课题组40报道了两例与Mn3CaO4立方烷结构相似的分子图16),该立方烷结构分别是由CO32-和SO32-固定。尽管配合物11和12的TON分别为96和117,氧气收率分别为19.2%和23.4%。但在光催化反应之后,通过DLS实验可以明显观察到纳米颗粒生成,表明该立方烷催化剂的氧化稳定性较差。

图16 配合物11 的结构示意图Fig. 16 Structural representation of complex 11.
综上所述,立方烷结构配合物金属中心上的水配体或者不饱和配位结构有利于水氧化的进行。以配合物1为例,其结构十分稳固,尽管Dismukes课题组最初报道其具有较好的水氧化活性,但Nocera等人的研究证实其光催化水氧化活性十分微弱,在Dismuke课题组使用的实验条件下,[Ru(bpy)3]3+无法将配合物1直接氧化,微弱的水氧化活性源于强氧化能力的与配合物1直接作用,该工作说明金属中心的水配体或者不饱和配位结构的重要性。一些立方烷结构的分子催化剂中具有配位水结构或是不饱和配位结构的金属中心是唯一的,例如配合物7a-7d,可以认定发生价态变化的金属中心一定是水配位的Co。但是对于更多的立方烷结构配合物而言,具有这样配位结构的金属中心通常至少有两个,而且是等价的。例如配合物6和8a活性中心只能归类于具有配位水结构或是不饱和配位结构一类的的金属中心,具体是哪一个原子,目前则无法判断,有待进一步深入研究。
2.2 铜为中心原子的配合物
如前文所述,钴基立方烷结构的分子催化剂被广泛应用于光催化水氧化28,36。相比之下,目前仅有一例Cu4O4分子催化剂被用于电催化水氧化41,而基于Cu4O4立方烷结构的光催化水氧化还未被进行研究。2018年,我们课题组首次42报道了一 例 八 核 铜 簇[Cu8(dpk·OH)8(OAc)4](ClO4)4(dpk·OH = 二(2-吡啶)酮)(配合物13,图17),该分子中包含两个结构相似的Cu4O4立方烷核,与Mn4CaO5簇中Mn3CaO4立方烷结构类似。最佳的O2收率为35.6%,TON为178,TOF为3.6 s-1,这些是目前报道的所有铜基光催化水氧化催化剂的最高值。我们认为该配合物具有高催化活性并不是因为配体的电子效应,而是因为邻近Cu(II)中心的配体上的羟基结构能在催化剂分子活性中心周围以氢键为作用力形成水分子网络结构,更有利于水分子在催化过程中对Cu(II)中心进攻,加速水氧化反应速率。一系列实验手段证实该双核Cu4O4是稳定的均相水氧化光催化剂,并没有形成CuOx等多相水氧化活性物种。

图17 配合物13 的球棍模型图Fig. 17 The ball-and-stick representation of complex 13.
该分子中仅存在一种类型的五配位结构的Cu中心,既然分子结构保持稳定,而六配位结构的Cu中心已经配位饱和,无法作为水进攻位点,因此可以认为分子结构中五配位Cu中心为活性位点。
3 基于全无机多金属氧酸盐的水氧化催化剂
多金属氧酸盐(Polyoxometalates,POMs),简称“多酸”,由高价态前过渡金属离子(主要指Mo、W、V、Nb、Ta)通过氧原子桥联形成的一类金属氧簇化合物43-45。首先,多酸作为抗氧化能力强的纯无机配体,可替代抗氧化能力较弱的有机配体来构筑过渡金属氧簇水氧化光催化剂。其次,多酸具有多个氧化还原活性的位点,能够进行快速的、可逆的以及逐步的多电子转移反应。第三,多酸具有明确的结构,其组成、能带结构、电荷、氧化还原性等均可进行调控。这些特性有助于设计与合成新型的光催化水氧化催化剂,从而从分子水平上模拟PSII中产氧中心Mn4CaO5簇。是一类非常有前景的水氧化催化剂46-49。
3.1 含钌的多金属氧酸盐水氧化催化剂
钌基多酸因其具有全无机配体,能在强氧化条件下保持稳定而不发生分解,备受人们的关注50-52。
2009 年, Hill 课 题 组53报 道 了 一 例[{Ru4O4(OH)2(H2O)4}(γ-SiW10O36)2]10-(Ru4-(SiW10)2,配合物14,图18),以[Ru(bpy)3]Cl2为光敏剂和Na2S2O8作为牺牲电子受体,在磷酸缓冲溶液中(pH 7.2),该催化剂获得了较高的TON为~350,TOF为~0.08 s-1,量子效率为~26%。
3.2 含钴的多金属氧酸盐水氧化催化剂
2014年,张志明,林文斌和王恩波等54系统地合成了四个基于十六核钴簇{Co16(PO4)4}的多酸基Co-Pi分子催化剂[{Co4(OH)3(PO4)}4(XW9O34)4]n-(X = Si, Ge, P, As)(配合物15a-15d,图19)。该结构中多酸杂原子可调变为Si、Ge、P、As元素,作者首次将杂原子为As和Ge的钴基多酸引入光催化水氧化中,且具有Co4O4立方烷单元,非常类似于产氧中心的Mn3CaO4核簇。该类纯无机化合物是基于地球丰产元素的稳定分子水氧化催化剂新结构模型,表现出较好的光催化产氧活性,并且杂原子是影响催化剂产氧活性的重要因素,其产氧活性顺序为15b > 15d > 15a > 15c。通过激光闪光光解实验、DLS、UV-Vis、31P NMR、POM萃取实验以及电感耦合等离子体质谱(ICP-MS)等实验证明以上四个不同杂原子结构相似的钴基多酸在光催化水氧化过程中是稳定的分子催化剂。此外,该多酸基Co-Pi分子为结构尚不明确的非晶态Co-Pi催化剂的模拟55,提供了一定的借鉴意义。
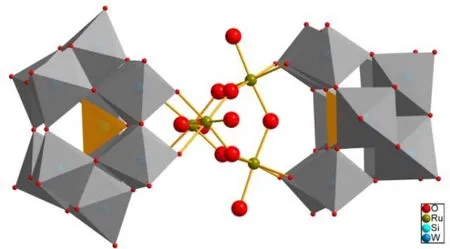
图18 14 的多面体和球棍结构模型Fig. 18 Polyhedral and ball-and-stick model of 14.

图19 15a-15d 的合成路线图54Fig. 19 The synthetic route to obtain {Co16(PO4)4}clusters 15a-15d.
2015年,我们课题组56报道了一例通过自组装形成的八核Co 基多酸水氧化催化剂[(A-a-SiW9O34)2Co8(OH)6(H2O)2(CO3)3]16-(配合物16,图核通过碳酸根连接起来,并位于两端的[A-α-SiW9O34]10-上,这是报道的第一例端基含有配体H2O的POM基催化剂。16在光催化水氧化体系中显示出非常好的产氧性能,其TON = 1436,TOF = 10 s-1,这是当时报道的基于廉价过渡金属催化剂中最高的TON值(表1)。包括UV-Vis、FT-IR、DFT、动态光散射、X射线光电子能谱(XPS)、电喷雾质谱(ESI-MS)、闪光光解等一系列实验证明16是一个稳定的、高效的水氧化催化剂。
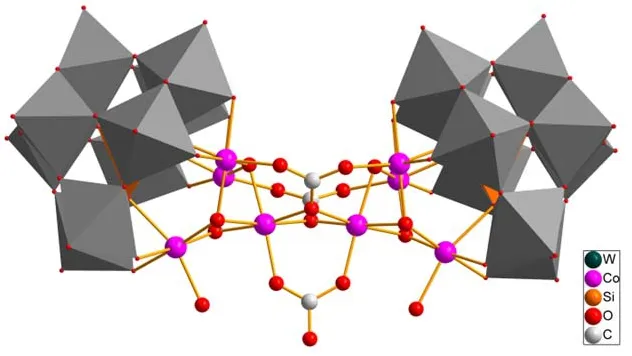
图20 16 的多面体和球棍结构模型Fig. 20 Polyhedral and ball-and-stick model of 16.
2016年,王新龙和苏忠民课题组57报道了首例含有Co-As核的光催化水氧化催化剂Na12[O9(OH)6}(A-a-SiW9O34)2]·8H2O(配合物17,图21),该结构包含了一个{AsO3}稳定的三齿配体,具有“融合”双准立方烷构型的多酸基,其TON = 115.2,TOF = 0.14 s-1,在相同的条件下,均高于之前报道的Co16(PO4)4(15c)的TON和TOF值,并与配合物16的TON和TOF值相当。作者通过陈化实验和动态光散射实验表明该基多酸在光水氧化过程中是稳定的催化剂。
3.3 含锰的多金属氧酸盐水氧化催化剂
2014年,Bonchio课题组58报道了首个含混合价态核的锰基多酸(CH3COO)3(A-a-SiW9O34)]6-(配合物18,图22),该结构与自然界PSII中的Mn4CaO5簇相似。该结构由MnIII3MnIVO3核,一个全无机的钨硅酸盐阴离子和三个有机醋酸配体组成的,为后续催化中心单电子氧化步骤提供了稳定和灵活的配位环境。以NaHCO3/Na2SiF6(pH 5.2)为缓冲溶液时,其量子效率达到1.7%,氧气收率为1.2%-3.7%,TON为5.2。而等量的MnSO4·H2O没有产氧活性,此外作者通过闪光光解实验研究了其氧化动力学过程。

图21 17 的多面体和球棍结构模型Fig. 21 Polyhedral and ball-and-stick model of 17.
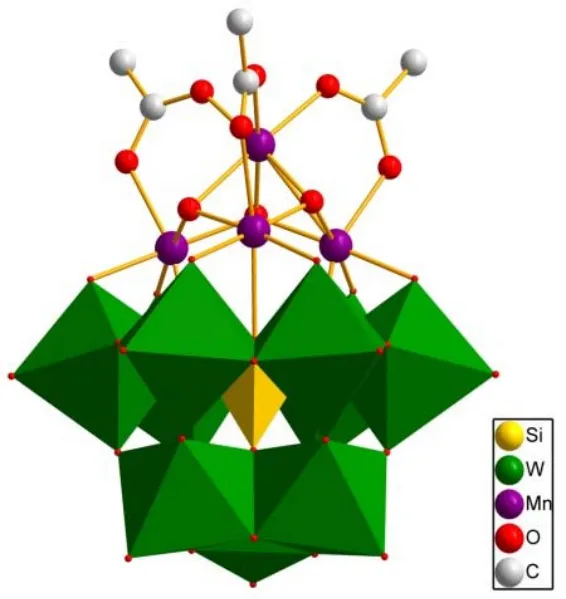
图22 18 的多面体和球棍结构模型Fig. 22 Polyhedral and ball-and-stick model of 18.
2016年,Streb课题组59报道了首例锰钒氧簇[Mn4V4O17(OAc)3]3-水 氧 化 光 催 化 剂(nBu4N)3[Mn4V4O17(OAc)3]·3H2O (配合物19,图23),由一个独特的三脚钒多酸[V4O13]6-和3个醋酸配体(OAc)构筑的混合价态的[MnIII2MnIV2O4]6+立方烷结构,对应于Kok循环中的S1态。单晶结构显示其Mn―O键的键长dMn―O为0.18-0.21 nm,与光系统PSII中dMn―O(OEC)≈ 0.18-0.2.0 nm相当。循环伏安测试和EPR测试表明在氧化条件下会发生两步连续的单电子氧化过程生成与Kok循环中S3态类似的作者考察了在[Ru(bpy)3]2+/Na2S2O8使用MeCN/H2O (9 : 1,v/v)作为溶剂的光催化水氧化体系,催化剂浓度为0.3 μmol·L-1时,催化活性最高,其TON和TOF高达1150和1.75 s-1(表1)。
3.4 含镍的多金属氧酸盐水氧化催化剂
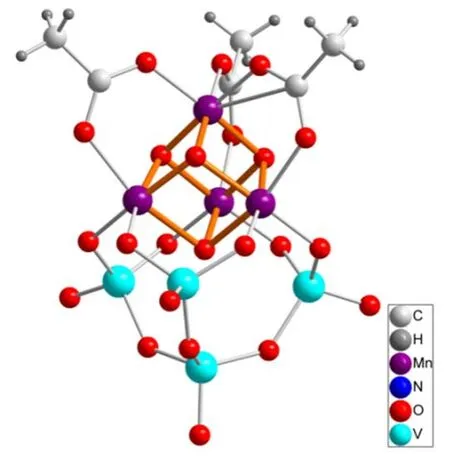
图23 19 的球棍模型Fig. 23 The ball-and-stick representation of 19.
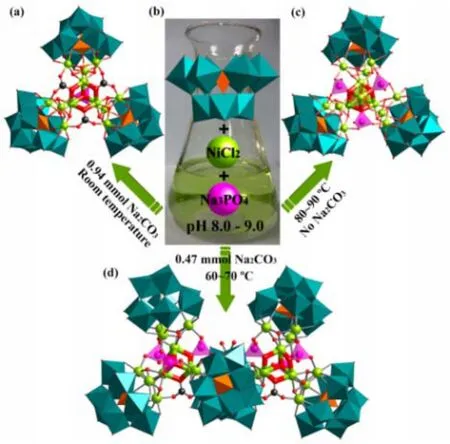
图24 多酸基高核镍簇20a-20c 的合成路线60Fig. 24 The synthetic route to obtain 20a-20c.
李阳光和王恩波课题组60随后报道了3个结构新颖的多酸基高核镍簇,分别为[Ni12(OH)9(CO3)3(PO4)(SiW9O34)3]24-({Ni12}) , [Ni13(H2O)3(OH)9(PO4)4(SiW9O34)3]25-({Ni13})以及[Ni25(H2O)2OH)18(CO3)2(PO4)6(SiW9O34)6]25-({Ni25})核(配合物20a-20c,图24)。该高核镍簇通过A-α-{SiW9O34}多酸单元将无机的{OH},{PO4}或{CO3}连接单元包裹在其中,均具有类似于植物叶绿素催化产氧中心{Mn4CaO5}的{Ni4O4}立方烷或{Ni3O3}准立方烷结构单元,显示了很好的可见光催化产氧活性。其{Ni12},{Ni13}和{Ni25}的TON分别为128.2,147.6和204.5 (表1),其中含有25个镍中心的多酸基高核镍簇{Ni25}是在此之前报道的活性最高的镍基光解水产氧催化剂。作者通过DLS、四庚基硝酸铵(THpANO3)甲苯萃取实验、UV-Vis光谱,毛细管电泳和陈化实验等共同验证了在光催化水氧化过程中3种多酸基高核镍簇是真正的均相催化剂,反应过程中没有分解。
4 多相体系的水氧化催化剂
近年来,基于第一过渡系金属的水氧化催化剂被广泛研究,目前大多数光催化水氧化体系都是以[Ru(bpy)3]2+或其衍生物为光敏剂,金属配合物或多酸作为分子催化剂,正在朝着与自然光合作用体系PSII多核Mn4CaO5反应中心的水氧化活性效率努力(表1)61。但由于[Ru(bpy)3]2+在水氧化过程中稳定性较差,随着长时间照射会发生不可逆的分解62,63,且吸收太阳光波长范围窄等缺点,寻找稳定性更好,捕光范围更宽的半导体材料作为捕光剂的水氧化体系被广泛关注64-67。以半导体BiVO4或氮化碳(PCN)为捕光剂,以立方烷助催化剂为体系也引起了研究者的广泛兴趣。
2016年,李斐课题组与李灿课题组几乎同时报道了以半导体BiVO4为捕光材料,钴基立方烷为分子催化剂,构筑了半导体与分子催化剂相耦合的杂合光催化水氧化体系。李斐课题组68将BiVO4与还原氧化石墨烯(RGO)耦合,立方烷钴基分子配合物Co4O4(O2CMe)4(py)4,(py = 吡啶),作为助催化剂(配合物1,图25),以AgNO3作为电子牺牲受体首次构建了半导体与分子催化剂相耦合的粉体光催化水氧化体系。RGO的引入有效地提高了BiVO4的光生电子和空穴的分离效率,其光催化产氧速率相比于无助催化剂条件下BiVO4提高了4倍。通过核磁等实验手段证实了该催化剂在反应过程中没有分解。
李灿课题组69以立方钴络合物为产氧助催化剂Co4O4(O2CMe)4L4(L = py (1), CNpy (1c), Mepy(1d)) (图26),以NaIO3作为电子牺牲受体,其中化合物1d的TOF = 2.0 s-1,1d/BiVO4的表观量子效率AQE为4.5%,是无助催化剂条件下BiVO4的9倍。同时首次利用了开尔文探针显微镜和表面光电压谱澄清带有合适配体的Co分子催化剂极大地增加了BiVO4半导体的光生载流子的分离能力。实验结果充分证明了光生空穴从半导体转移到分子催化剂的可行性,这为后续研究高效的分子催化剂和适合水氧化的半导体材料提供了新的思路。
2018年,王心晨课题组70将“Co4O4”立方烷Co4O4(O2CMe)4L4(L = py (1), CNpy (1c), Mepy(1d), Brpy (1e), OMepy (1g)作为均相助催化剂调控氮化碳骨架(图27),不仅增加了两者的有效接触,同时也为反应提供了更多的活性位点。以AgNO3作为电子牺牲受体,La2O3(0.2 g)作为缓冲溶液以及“Co4O4(1)”的负载量为3.0% (w)时,相比单纯的氮化碳,其光催化产氧活性提高了19倍。

图25 BiVO4-RGO 与钴基立方烷催化剂1 耦合光催化水氧化体系示意图68Fig. 25 The photocatalytic O2 evolution by a BiVO4-RGO/1 hybrid system 68.

图26 BiVO4 与钻基立方烷催化剂(1,1c,1d)耦合光催化水氧化体系示意图69Fig. 26 The photosynthetic system composed of BiVO4, molecular Co catalysts (1, 1c, 1d) 69.

图27 PCN 与Co4O4 立方烷耦合光催化水氧化体系示意图70Fig. 27 The photocatalytic O2 evolution by a PCN/Co4O4 hybrid system 70.
5 结论
综上所述,过渡金属立方烷结构的分子催化剂在光驱动水氧化反应中已取得了一定的进展(表1) 。无论是以[Ru(bpy)3]2+为光敏剂还是以半导体为捕光材料,合理设计以M4O4(M = Mn, Co, Ni,Cu或其他金属)为中心的立方烷或准立方烷结构的分子催化剂具有极其重要的意义。根据以上对各种立方烷结构的光催化水氧化催化剂的介绍总结可以发现:(1)催化剂稳定性与配体的稳定性、金属中心(到目前为止,相比于钴基配合物而言,稳定的基于有机配体的镍基配合物的水氧化光催化剂还未见报道)以及配位环境有关。(2)在研究立方烷配合物催化的光催化水氧化反应时,尤其是钴基立方烷催化剂,必须保证催化剂的纯度,防止合成过程中金属离子杂质的引入,这对催化剂活性的甄别是极其重要的,也是必须的。(3)通过调节这些分子催化剂的活性金属中心以及改变外围配体或末端官能基团以实现其氧化还原电位和电子结构的调变。(4)在以半导体为捕光材料的体系中,立方烷分子反应前后的稳定性需要考虑。(5)通过更加合理设计实验与表征手段,对催化反应过程进行跟踪表征,为催化剂机理方面提供理论和实验指导。
总之,从分子层面上理解光系统PSII的催化机理,可为设计具有高效稳定的廉价立方烷基水氧化催化剂提供一定的理论依据。从长远的角度来看,基于立方烷结构的分子催化剂在光催化水氧化中的研究仍是一个非常有潜力的研究方向。而且具有重要的应用价值,同时也是极具挑战性的重要科学问题。
