光催化全解水助催化剂的设计与构建
2020-03-27孙尚聪张旭雅刘显龙潘伦张香文邹吉军
孙尚聪,张旭雅,刘显龙,潘伦,2,张香文,2,邹吉军,2,*
1 天津大学化工学院, 绿色合成与转化教育部重点实验室,天津 300072
2 天津化学化工协同创新中心, 天津 300072
1 引言
随着经济的高速发展和人口数量的不断增多,人类社会发展对能源的依赖程度与日俱增,传统化石能源的快速消耗引发了严峻的能源危机,并导致严重的环境污染和生态破坏1-6。开发利用清洁可再生的绿色能源越来越迫在眉睫。太阳能具有储量丰富、分布广泛、无污染等优势,被视为一种理想的可再生能源形式5,7-11。但太阳能存在能流密度低、时空分布不连续等问题,需要合理的能量转化方式对其有效存储和利用。光催化水分解制氢相较于其他制氢方法更加温和有效,得到人们的广泛关注4,12-17。
光催化水分解主要包括三个基元反应步骤:(I)半导体催化剂吸收光和产生光生电荷;(II)光生电荷(电子和空穴)的分离及迁移;(III)表面氧化还原反应发生18-20。半导体对光的吸收能力(步骤I)主要由其自身的禁带宽度所决定,而电荷分离和表面反应过程通常都需要通过助催化剂来加以促进,进而提高光催化反应的整体效率21-23。如图1所示,对于产氢反应而言,半导体产生的光生电子的还原电势需要负于0 V (vsNHE) (pH = 0),而产氧过程则要求光生空穴的氧化电势高于1.23 V (vsNHE)。
现阶段的研究大多集中在如何促进光催化水分解产氢与产氧半反应,但半反应中使用牺牲剂所带来的成本负担使其难以产生实际经济效益1,24-27,实现光催化纯水分解产氢及产氧也就变得尤为重要。但是,现阶段光催化全解水却受到光生电荷分离效率低、表面反应速率慢以及氢氧复合逆反应严重等因素的制约28-30,需要引入助催化剂来调节半导体的电荷分离及表面反应性能。尤其地,全解水中使用的助催化剂需要选择性地催化H2或O2的生成,同时抑制H2和O2复合的逆反应31,32。本文将对助催化剂在光催化反应中的重要作用以及目前常用的助催化剂进行分析和介绍,对光催化全解水过程中双助催化剂体系的构建及作用机理进行总结,并根据限制全解水的关键因素提出新型助催化剂的设计策略。

图1 光催化的基本过程Fig. 1 Basic process of photocatalysis.
2 助催化剂在光催化分解水中的作用
助催化剂对提高光催化剂的活性及稳定性等方面起着至关重要的作用。尽管助催化剂本身通常并不具有光吸收性能,将其与半导体材料耦合后却可以使光催化活性得到显著提高,作用机理主要包括以下几个方面33-36:
(1)提高主体光催化剂界面处的电子空穴分离效率。如图1所示,半导体导带上的光生电子可以定向转移到还原助催化剂上并参与还原反应,而价带上的光生空穴能够定向转移到氧化助催化剂上并参与氧化反应。保证半导体和助催化剂之间的紧密接触(欧姆接触、肖特基接触等)是提高电荷分离效率的关键因素。通常情况下,由于半导体和助催化剂之间功函数的差异,二者界面处的能带会发生弯曲并形成空间电荷层,进而促进半导体上光生电荷的迁移,抑制电子和空穴的体相复合37。
(2)降低半导体表面H2和O2生成反应的活化能或过电势并提供反应活性位点。相对于产氢半反应,产氧过程通常具有较大的过电势(~700 mV),其中O―O键形成常被认为是速控步骤38。研究表明,通过构建合适的助催化剂可以有效降低水分解产氧的活化能39。通过对助催化剂的形貌、尺寸、含量进行调控,可以提供丰富的活性位点以提高表面反应速率。
(3)抑制光腐蚀并提高光催化剂稳定性。半导体产生的电子或空穴不仅可以参与水的氧化还原过程,还可能与自身发生反应并导致光腐蚀。许多可见光响应的硫(氧)化物和氮(氧)化物等均易被空穴氧化分解。而助催化剂可以通过促进电荷分离和表面反应及时迁移走半导体内部的光生电荷,抑制光腐蚀的发生。也有研究表明,助催化剂可以减少半导体自身的表面捕获态,提高其稳定性40。
(4)抑制逆反应发生。对于水分解,除了要加快正反应速率以促进氢氧产生之外,抑制二者的复合也十分重要。常见的助催化剂如Pt等在促进氢气产生的同时也会催化氢氧复合,限制整体反应效率。通过调控助催化剂的化学状态可以有效抑制逆反应的发生41,42。例如氧化态的PtOx比单质Pt具有更高的产氢速率和更强的抑制氢氧复合的能力30。
同时,助催化剂的负载量也会影响其性能。光催化剂的活性通常先随助催化剂负载量的增大而逐渐提高,达到最大值后进一步增大助催化剂的负载量反而会使活性下降,呈火山图规律变化,其原因如下:
在起始阶段,提高助催化剂负载量可以通过促进电荷分离,增多表面活性位点以提高反应活性;但当负载量进一步提高时,多余的助催化剂会遮盖半导体表面吸光位点,使光吸收能力下降并抑制光生电荷产生,也会成为新的电荷复合中心,加快逆反应而限制整体反应效率。
3 用于光催化水分解的助催化剂
从本质上说,水分子在活性中心的吸附、解离过程是水分解反应的起始步骤,是决定光催化效率的关键因素之一。由于其本质上与电催化类似,许多电催化剂均可作为光催化助催化剂参与反应,并取得了良好的效果43-48。
3.1 产氢助催化剂
贵金属如Pt、Rh等具有更大的功函数和更低的费米能级,易与半导体形成肖特基势垒并作为优良的电子捕获陷阱参与析氢反应。相比于单一的TiO2、C3N4等半导体催化剂,当添加少量(通常为质量分数1%-3%)的贵金属助催化剂时,相同条件下的光催化产氢活性可以得到数量级式的提升。但是贵金属的含量稀少、使用成本过高,开发替代型非贵金属助催化剂日益受到重视。例如,一些低成本的过渡金属单质Co、Ni和Cu等,它们的作用机理通常与贵金属类似,即形成金属/半导体肖特基势垒以促进电子转移,而且能够催化质子还原成H2分子49-51。Sun等将超细的Ni金属单质负载于CdS纳米片上,使光催化产氢活性明显提升,并实现了糠醇和5-羟甲基糠醛氧化反应的耦合49。Lin等报道,负载Cu的介孔Nb2O5上的H2生成速率大约是纯Nb2O5的5倍52。
过渡金属氧化物或氢氧化物也具有产氢催化活性,包括NiO、Ni(OH)2、CuO、Cu2O等。Gu等发现,NiO可以提高C3N4的光催化产氢活性。在可见光照射下,产氢速率是纯C3N4的近430倍,其原因是NiO和C3N4通过C―O―Ni键形成相互作用,促进了光生电子从C3N4到NiO上的转移53。Niu等发现Ni(OH)2作为助催化剂使ZnxCd1-xS的产氢活性明显提高54。一般而言,金属氧化物和氢氧化物的电势E0(MOx/M, M为金属)或E0(M(OH)x/M)通常位于半导体的导带底和产氢电势E(H+/H2, 0 eV)之间,因此会首先被电子部分还原成金属单质进而参与产氢反应。Foo等证明负载于TiO2上的Cu2O助催化剂在光照下会首先被光生电子还原成金属Cu,后者作为活性位点参与产氢反应55。
过渡金属硫化物和磷化物也常被用作产氢助催化剂,如NiS、NiPx和MoS2等56,57。以Ni2P为例,由于其具有类似贵金属Pt的电子性质,使TiO2、C3N4和CdS等半导体的光催化产氢活性明显提高58-60。MoS2是一种优异的电催化析氢催化剂,其二维结构边缘位置具有丰富的配位不饱和结构,提供了大量的活性位点。近来,MoS2也被用作一种高效的光催化助催化剂,表现出甚至优于贵金属的催化活性。Ye等提出了一种简便的离心液相剥离法,制备具有纳米级横向尺寸的单层强化MoS2纳米片(单层产率36%)并将其负载于CdS上,产氢活性达到5.8 mmol·h-1,在420 nm处的表观量子效率达到77.2%61。另外。其他材料如碳化钨、碳量子点、还原氧化石墨烯等也是常用的产氢助催化剂62。
3.2 产氧助催化剂
水分解产氧需要较高的反应活化能并且涉及到四电子转移,通常被认为是全解水的瓶颈,助催化剂在产氧反应中通常是不可或缺的31。常用的光催化产氧助催化剂以贵金属和过渡金属氧化物为主。贵金属氧化物主要有RuO2和IrO2等,它们在大部分产氧反应中表现出优异的性能,但是除了价格昂贵之外,RuO2在碱性溶液中的抗腐蚀能力较弱,而IrO2的导电性较差,极大限制了产氧效率的提高。过渡金属Co、Ni、Mn、Fe等的氧化物及氢氧化物等也常被用作产氧助催化剂,它们的价格相对低廉,且具有优良的抗化学和光腐蚀性能40,63。例如,CoOx可以降低产氧反应的活化能并与主体催化剂形成内建电场来促进电荷的转移64。Wang等设计Co3O4量子点为产氧助催化剂,通过精确调控助催化剂的负载量和样品焙烧温度,有效提高了C3N4上的电荷分离和传输效率,使其光催化产氧活性比纯C3N4提高了4倍(图2)65。王心晨等将Co(OH)2助催化剂负载于C3N4上,在促进电子转移的同时降低了O―O键形成的能垒,进而提高产氧反应动力学速率,在紫外可见(λ> 300 nm)和纯可见光照射(λ> 420 nm)下的光催化产氧速率分别达到27.4和7.1 μmol·h-166。

图2 (a)在300 °C 下处理的负载不同比例的量子点Co3O4 的C3N4 的光催化产氧活性;(b) Co3O4-C3N4 在焙烧不同温度时的光催化产氧活性;(c)电化学阻抗谱(EIS)曲线和(d)可见光照射下的光电流响应曲线Fig. 2 (a) Amount of O2 evolution for C3N4 modified with different ratios of Co3O4 QDs treated at 300 °C,(b) amount of O2 evolution for Co3O4-C3N4 annealed at different temperatures, (c) electrochemical impedance spectroscopy (EIS) plots and d) periodic on/off photocurrent response under visible-light irradiations.
一些双金属氧化物或氢氧化物也能显著提高产氧反应活性。相对于单一的金属氧化物或氢氧化物,双金属化合物中二元金属的协同作用可以调控助催化剂的电子结构,增强对水分子的活化能力及促进O―O键的形成。王心晨等制备MnCo2O4尖晶石作为产氧助催化剂,获得比纯Co和纯Mn化合物更加优异的性能,尖晶石中Co、Mn混合价态的协同作用有效地提高了产氧反应动力学67。Teranish等将Co掺杂进Mn3O4中并将其负载于Rh@Cr2O3/SrTiO3上,获得良好的活性并实现了氢氧产出2 : 1的全解水反应,对比实验表明,CoxMn1-xO4作为助催化剂的性能是纯Mn3O4的1.8倍68。
金属磷化物和磷酸盐均可作为优良的空穴传输层以提高光催化产氧活性。Wu等制备了质子化的C3N4用于光催化产氧,当添加磷酸钴盐(CoPi)为助催化剂时,其活性明显提高69。类似的,将CoPi负载于BiVO4上时,产氧活性随着负载量呈火山图规律变化,当CoPi的负载量为1%时,产氧速率达到纯BiVO4的6.8倍70。另外,新型产氧助催化剂也在不断的开发之中,如CaI、B2O3-xNx以及一些过渡金属配合物等,石墨烯和还原氧化石墨烯也被报道作为氧气生成的活性位点40。
3.3 用于光催化全解水的双助催化剂体系
光催化产氢和产氧半反应的效率在不断提高,在添加Na2S-Na2SO3为牺牲剂时,Pt-PdS/CdS在420 nm处的表观产氢量子效率达到了93%,是目前文献报道的最高值之一71。然而,不添加任何牺牲剂或pH调节剂的光催化纯水分解具有更大的意义,也更加困难。一方面,纯水中半导体上的电荷复合速率相对较快,极大地抑制了全解水效率。另一方面,一个稳定光催化体系不仅要求2 : 1的氢气和氧气产出,而且需要有效地抑制氢氧复合的逆反应以提高整体效率。
双助催化剂负载是全解水反应最常用策略之一,要求选择合适的氧化和还原助催化剂并将二者分别负载于半导体上,通过进一步调控助催化剂的含量、尺寸、形貌和分布状态等参数促进电荷的空间分离,并调控氢气和氧气的生成速率。目前大部分的全解水体系都有赖于双助催化剂的辅助。双助催化剂体系最早由Domen课题组报道,通过在(Ga1-xZnx)(N1-xOx)负载具有核壳结构的Rh-Cr2O3复合结构和Mn3O4纳米颗粒,二者分别作为产氢和产氧反应的助催化剂,光电测试分别证实了两种助催化剂在水分解反应中的作用,如图3所示。相对于仅负载Rh-Cr2O3助催化剂的体系,双助催化剂使全解水活性提高1倍多,并保证了优良的稳定性。特别的,由于Rh-Cr2O3中无定形的Cr2O3壳层对质子具有选择透过性而能阻止O2透过,有效抑制了氢氧复合逆反应的发生72。
同样的策略也被用于d0型半导体。Domen等采用RuOx/Cr2O3为产氢助催化剂,IrO2作为产氧助催化剂,实现了ZrO2/TaON体系的全解水。结果表明,相对于仅有产氢助催化剂体系,双助催化剂体系能够实现更加高效和稳定的水分解反应8,73。双助催化剂也可以促进Z型体系的全解水反应。Li等采用Pt为产氢助催化剂、PtOx为产氧助催化剂,以MgTa2O6-xNy/TaON-(IO3-/I-)-WO3为光催化剂,其中,Pt/MgTa2O6-xNy/TaON构成产氢活性组分,PtOx/WO3为产氧组分,实现了在420 nm处6.8%的表观量子效率74。进一步地,Domen课题组将双助催化剂用于三元全固态Z 型全解水体系,以Rh/SrTiO3(SrTiO3:La,Rh)为产氢光催化组分,以Rh/BiVO4(BiVO4:Mo)为产氧活性组分,采用Au层为固体电子媒介,在419 nm处的表观量子效率达到了30%,且太阳能转换效率(STH)达到1.1%,是目前文献报道的最高值之一(如图4所示)75。

图3 可见光驱动的用Mn3O4 和Rh/Cr2O3(核/壳)纳米粒子改性的GaN:ZnO 固溶体的光解水机理示意图Fig. 3 A proposed reaction mechanism for visible-lightdriven overall water splitting on GaN:ZnO modified with Mn3O4 and Rh/Cr2O3 (core/shell) nanoparticles.
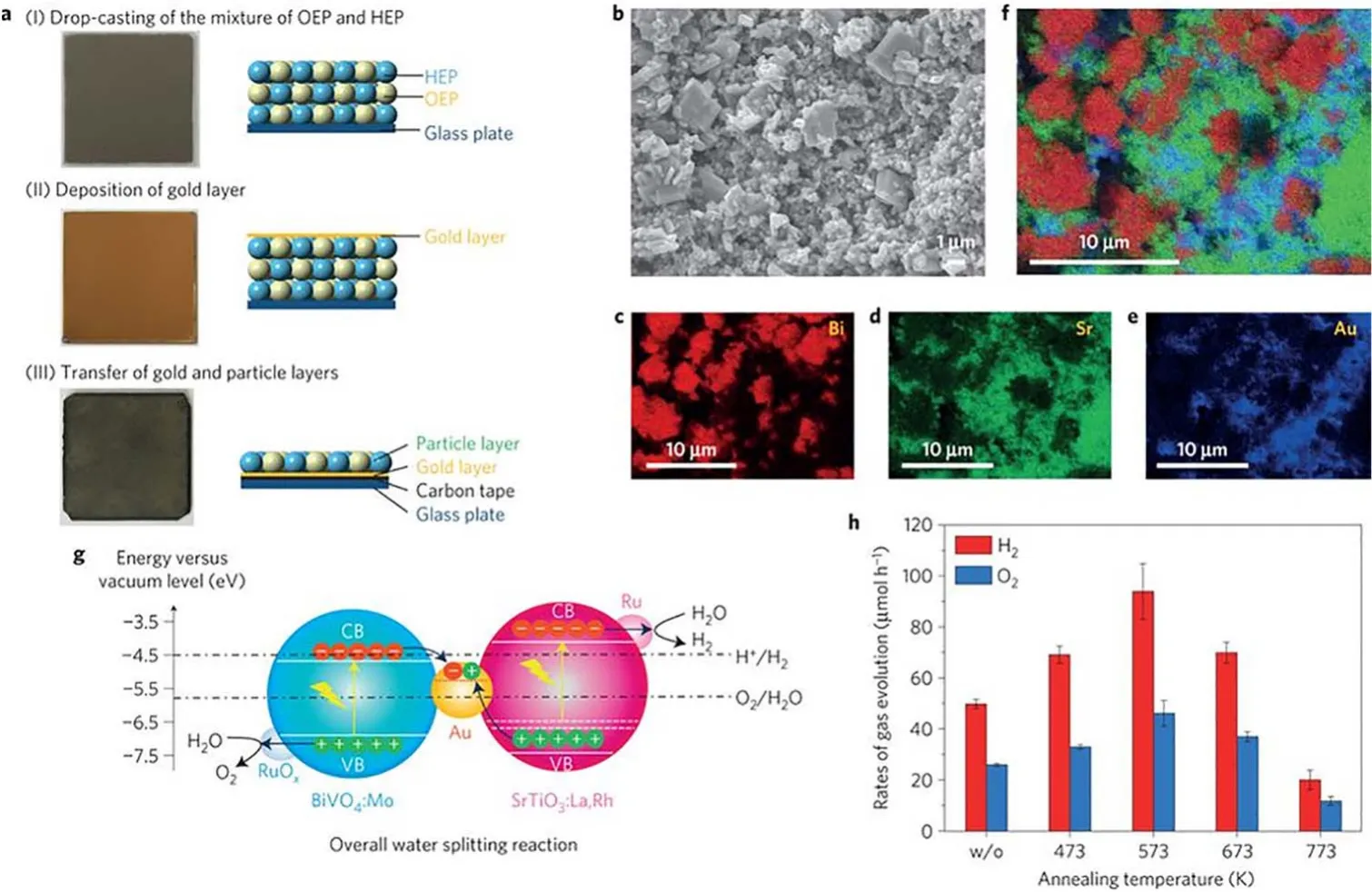
图4 颗粒转移法制备的SrTiO3:La,Rh/Au/BiVO4:Mo 催化剂Fig. 4 SrTiO3:La,Rh/Au/BiVO4:Mo sheet prepared by particle transfer method.
具有大共轭结构的非金属聚合物光催化剂发展快速76,77。以氮化碳(C3N4)为例,它的电子结构和形貌易于调控,适于水分解的能带结构(禁带宽度2.7 eV,导带低位置在-1.0 eV,价带顶位置在+1.7 eV左右),且制备方法简便、成本低廉78-80。C3N4在光催化产氢或产氧反应、污染物降解、分子氧活化等领域得到广泛关注,其全解水应用也逐渐得到了重视69,81-83。王心晨等制备了Pt/C3N4光催化剂(3% Pt)并实现了氢氧化学计量比产出的全解水反应,XPS等结果表明,催化剂中的Pt以混合价态形式存在,其中金属单质型Pt作为还原活性位点促进氢气产生,而氧化态的PtOx作为产氧活性位。当进一步添加CoOx为产氧助催化剂时, Pt-CoOx/C3N4催化剂在300 W氙灯(λ> 420 nm)的光照条件下达到了1.2和0.6 μmol·h-1的氢氧产出,这也是在聚合物半导体体系实现光催化全解水的首次报道84。此后,他们以Pt作为C3N4上产氢半反应的助催化剂,同时引入CoP作为产氧助催化剂。结果发现,CoP的引入有效降低了产氧半反应的能垒,使产氧活性提高了数倍;同时,由于Pt与CoP两种助催化剂的协同作用,光生电子选择性向Pt迁移,光生空穴向CoP迁移,大大提高了光生电荷的分离效率,Pt-CoP/C3N4的全解水活性比仅引入Pt或者CoP都有显著提高,也表现出了优异的稳定性(如图5所示)85。
上述结果充分证实双助催化剂策略在光催化全解水中的有效性,但仍需要对助催化剂的种类、含量以及空间位点进行调控。由于全解水面临着电荷分离效率低、逆反应速率快等限制,在单一半导体上实现氧化和还原反应活性位点的空间分离是提高光催化效率的重要方式。如图6所示,王心晨等利用SiO2为模板剂制备了双面的氮化碳(Co3O4/HCNS/Pt)空心结构,将还原助催化剂Pt沉积在空心球的内表面而将氧化助催化剂Co3O4负载到空心球的外表面,实现H2和O2生成位点的空间分离,相对于Co3O4和Pt均分布于外表面的(Co3O4+ Pt)/HCNS光催化剂,Co3O4/HCNS/Pt具有更高的全解水活性86。
此外,通过合理调控助催化剂与半导体的接触方式如构建0D/2D复合体系可有效提高其活性。将0D型助催化剂负载于2D半导体上,在保持原有催化剂光吸收能力的同时丰富了表面活性位点和电荷分离中心的数量。前述Pt/C3N4, Pt-CoP/C3N4等均取得了良好的全解水活性84,85,证实了0D/2D体系的优势。
3.4 强化水分子活化的复合助催化剂
水分子在催化剂表面的活化(吸附解离)是水分解反应的起始和关键步骤。常规的产氢助催化剂如Pt等虽然具有良好的吸附质子和促进质子复合成氢气的能力,但对水分子吸附能力较差;而Co、Mn、Ni氢氧化物等氧化助催化剂难以有效地促进质子复合。在常规双助催化剂修饰的半导体中,这两种助催化剂通常是空间分离的,难以同时实现对水分子活化和氢氧产生的促进。因此全解水中水分子活化限制了反应效率。设计新型的复合助催化剂体系以加速水分解的表面反应动力学尤为重要。

图5 (A)氙灯的全光谱照射下负载不同助催化剂的C3N4 的光催化全解水活性对比(pH = 3,反应时间4 h)(B)全光谱下使用Pt (3%)-CoP (3%)/C3N4 的全解水稳定性测试结果Fig. 5 (A) Photocatalytic overall water splitting using C3N4 loaded with different cocatalysts under full arc irradiation of a Xe lamp at pH = 3 for 4 h, and (B) overall water splitting by Pt (3%)-CoP (3%)/C3N4.

图6 (a) Co3O4/HCNS/Pt 和(b) (Co3O4 + Pt)/HCNS 全光谱(λ > 300 nm)下光催化全解水的活性对比86Fig. 6 Time courses of photocatalytic evolution of H2 and O2 using (a) Co3O4/HCNS/Pt and(b) (Co3O4 + Pt)/HCNS under UV irradiation (λ > 300 nm) 86.
本课题组设计一种具有强协同作用的复合助催化剂Pt/Ni(OH)2,利用原位光诱导策略,将Ni2P前驱体光驱动氧化为Ni(OH)2并原位生长于C3N4纳米片上,之后利用静电相互作用将Pt以小团簇的形式选择性地沉积到Ni(OH)2的表面,形成Pt/Ni(OH)2复合助催化剂,如图7所示。理论计算和实验结果表明,纳米Pt与Ni(OH)2的紧密接触引发了强Ptσ+-Oσ--Niσ+交互作用,这种交互作用在强化电子传递的同时,增强了催化剂在表面反应过程中对H2O分子的吸附能力,同时使H―OH键更易被活化,有效降低了H2O分子解离的活化能,进而提高水分解过程中的表面反应动力学速率。将Pt/Ni(OH)2复合助催化剂负载在C3N4上,活性比单一的Pt 或Ni(OH)2助催化剂均明显提高,Pt/Ni(OH)2-C3N4在420 nm处的光催化全解水的表观量子效率达到了1.8%。相较于Pt、Ni(OH)2分别负载在C3N4上的常规双助催化剂体系而言,Pt/Ni(OH)2也具有明显的优势。值得一提的是,Pt/Ni(OH)2作为助催化剂实现了TiO2上的全解水反应,而常规的双助催化剂策略并不能使TiO2全解水87。

图7 (a) Pt/Ni(OH)2-C3N4 的制备流程示意图,(b) C3N4 和(c) TiO2 基半导体负载不同助催化剂时3 h 内的产氢半反应和全解水活性对比Fig. 7 (a) Schematic diagram of the preparation of Pt/Ni(OH)2-C3N4; averaged photocatalytic hydrogen evolution reaction(HER) and overall water splitting rate in 3 h of different cocatalysts loaded (b) C3N4 and (c) TiO2.
4 结论与展望
光催化水分解可以将太阳能转化为氢能并加以存储和利用。光催化产氢与产氧半反应的研究得到很大发展,但在不添加任何牺牲剂条件下全解水更具前景,也更具挑战。采用助催化剂对半导体表面进行修饰是提高光催化剂效率的有效手段,它可以促进半导体上光生电荷的分离和迁移,降低反应的活化能,为水分解反应提供活性位点,抑制逆反应并提高光催化剂的耐光腐蚀能力和稳定性。常用的还原助催化剂主要有贵金属(如Pt、Au等)、过渡金属及其磷化物或硫化物等;而产氧助催化剂主要包括贵金属氧化物(如RuO2、IrO2等)、过渡金属Co、Ni、Fe、Mn等的氧化物和氢氧化物。采用双助催化剂修饰半导体是实现光催化全解水的主要策略,有助于在促进半导体上电荷分离的同时分别提供氢氧产生的活性位点。进一步地,采用复合助催化剂促进水分子活化以提高光催化反应中的表面动力学,有利于实现更高效的全解水过程。
综上所述,助催化剂在光催化水分解领域有着至关重要的作用,目前助催化剂的性能仍然有待提升,对助催化剂的精确调控和深入的机理解释尚待完善,主要发展方向包括以下几个方面:
(1)构建具有多组分、多功能的复合助催化剂,在提供产氢产氧活性位点、促进电荷分离的同时,增强对水分子的活化(吸附、解离)能力,通过促进表面反应动力学来提高整体效率;
(2)对助催化剂的形貌、结构和缺陷及配位状态等参数进行更加精确的调控,强化助催化剂与半导体催化剂的相互作用,优化助催化剂的组成和化学状态以抑制氢氧复合逆反应;
(3)从本质上看,光催化表面反应与电催化反应有着极大相似性和类比性,因此可借鉴电催化水分解的催化剂设计策略来构筑光催化水分解的助催化剂。
