大位阻苊基α-二亚胺镍催化剂的制备及催化乙烯均聚
2020-03-26侯彦辉崔咪咪李景民
侯彦辉,孟 浩,崔咪咪,李景民,杨 敏
(1.天津工业大学 材料科学与工程学院,天津 300387;2.天津工业大学 省部共建分离膜与膜过程国家重点实验室,天津 300387;3.河北工业大学 化工学院,天津 300130)
聚烯烃工业的发展象征着一个国家的石油化学工业的发展,其中聚乙烯是通用合成树脂中应用最广泛的品种[1]。聚烯烃技术的关键是催化剂,聚烯烃性能的提高与聚烯烃催化剂的发展有着密切的联系。在历经Ziegler-Natta 催化剂和茂金属催化剂在聚烯烃领域的应用之后,1995 年,Brookhart 等[2]发现含有大体积的α-二亚胺的镍、钯的配合物对烯烃聚合具有良好的催化作用,才由此引发了对后过渡金属催化剂研发的热潮[3-4]。相较于应用较早的两类催化剂,后过渡金属催化剂优势明显:成本低廉,既能够催化单一烯烃单体聚合得到高支化或超支化的产物,也能够催化烯烃与极性单体的共聚,通过分子水平上的定向控制实现目标聚合物的共聚[5-9],具有高亲电性的镍、钯阳离子活性中心,有利于烯烃快速的配位插入,催化活性高,同时也具有一定的稳定性,对水和空气表现出较低的敏感性[10-14]。
α-二亚胺后过渡金属催化剂通常表现出的活性较高,但耐热性差的缺陷大大限制了它在工业中的应用,因此改变配合物的结构以提高其热稳定性成为许多人研究的目标[15-17]。Guan 等[18]设计了一个基于环番的α-二亚胺配体催化剂,该种催化剂在高温条件下刚性环仍然可以实现固定苯环,抑制相应的链转移和取代基的活化过程,表现出较高的催化活性[19-20]。Rhinehart 等[21-22]设计出的具有大位阻基团的α-二亚胺催化剂热稳定性优异,能够在100 ℃催化乙烯表现出较高的活性。Liu 等[23]设计合成了一系列不同螯合环大小的二亚胺配体,探究了其对催化活性的影响。Feldman 等[24]同时设计出了基于樟脑醌结构具有大位阻基团的α-二亚胺催化剂,研究表明碳骨架位阻较大能够提高催化剂活性及热稳定性。本课题组近年一直致力于α-二亚胺镍催化剂的研究,侯彦辉等[25]制备的具有大位阻基团的α-二亚胺镍催化剂能够在70 ℃实现对乙烯的均聚,并保持较高的活性。杨敏等[26]通过调整苊醌骨架结构,引入大位阻基团,实现较高温度条件下的高活性催化。当配体骨架上的取代基增大时,有利于抑制β-H 的消除[27],因此认为,取代基的增大有利于活性及热稳定性的提高。基于以上研究,本文通过调整配体结构,对位引入苯环和羟基基团实现更大空间位阻,以一氯二乙基铝作助催化剂对乙烯进行催化聚合,探究催化性能及不同条件下对聚合物结构的影响。
1 实验部分
1.1 原料与设备
原料:二氯甲烷(分析纯,经氢化钙干燥后使用)、甲苯(分析纯),天津市风船化学试剂科技有限公司产品;正己烷(分析纯),天津光复科技有限公司产品;四氢呋喃(分析纯),天津市风船化学试剂科技有限公司产品;以上后3 种试剂均在氩气保护下加入金属钠回流,使用前蒸出。无水乙醇、石油醚、乙酸乙酯,均为分析纯,天津恒山化工科技有限公司产品;苊醌,南京康满林化工实业有限公司产品;对甲基苯磺酸、三苯基磷钯,上海泰坦科技有限公司产品;2,6-二异丙基苯胺、一氯二乙基铝(DEAC),1.0 mol/L 正己烷溶液,百灵威科有限公司产品;4-羟甲基苯硼酸,安耐吉化学有限公司产品;碳酸钾,天津市风船化学试剂科技有限公司产品;乙烯,聚合级,氩气,空气化工(天津)有限公司产品。
设备:AVANGE-400M 型核磁共振波谱仪,德国Bruker 公司产品;Nicolet-iS50 型傅里叶红外光谱仪,德国赛默飞世尔科技公司产品;RW-10 型旋转蒸发仪,德国IKA 公司产品;DSC200F3 型差示扫描量热仪,德国耐驰公司产品;Varian 7.0T FTMS 型质谱仪,美国Varian 公司产品。
1.2 配体及催化剂的制备
1.2.1 4-溴-2,6-二异丙基苯胺(A)
取250 mL 干燥洁净的茄形瓶,依次加入2,6-二异丙基苯胺(22.2 mL,117.7 mmol)和 100 mL 混合溶剂(V(二氯甲烷)∶V(乙醇)=1 ∶1),在冰浴下搅拌的过程中逐滴加入液溴(7 mL,136.6 mmol),滴加完毕后撤掉冰浴,室温下搅拌过夜。反应结束后,减压蒸出部分溶剂,加入乙酸乙酯,静置片刻后析出大量固体,用乙酸乙酯抽滤,将收集到的固体继续用乙酸乙酯洗涤,以去除过量残余的液溴,再次抽滤,得白色纯净产物,产率93%。
1.2.2 N,N-二(2,6-二异丙基-4-溴)苊烯-1,2-二亚胺(B)
取250 mL 干燥洁净的茄型瓶,依次加入苊醌(1.0 g,5.5 mmol)和 4-溴-2,6-二异丙基苯胺(3.6 g,14.0 mmol),少量对甲基苯磺酸作催化剂,适量甲苯作溶剂,加热回流6 h,颜色为橙色,TLC 监测反应进度,反应完成后将甲苯蒸出,得到的粗产物用二氯甲烷溶解,通过柱层析纯化,产率为81%。
1.2.3 N,N-二(2,6-二异丙基-4-羟甲基苯基)苊烯-1,2-二亚胺(L)
取100 mL 干燥洁净的茄型瓶,称取化合物B(0.2 g,0.3 mmol)、对羟甲基苯硼酸(0.12 g,0.8 mmol)和无水碳酸钾(0.25 g,0.18 mmol),加入 THF 30 mL 和蒸馏水18 mL,之后加入少量三苯基膦钯作催化剂,用氩气置换气体,之后快速升温加热回流10 h,溶液为橙红色,停止加热后,溶液分层,用二氯甲烷进行萃取,取有机相加入无水硫酸钠干燥,之后抽滤浓缩,将粗产物通过柱层析纯化,得橙红色纯净目标产物,产率为78%。1H NMR(400 MHz,CDCl3)δ 7.90(d,J = 8.5 Hz,2H),7.75(d,J = 7.5 Hz,4H),7.58 -7.45(m,8H),7.44 -7.34(m,4H),4.79(s,4H),3.10(s,4H),1.30(d,J=6.4 Hz,12H),1.04(d,J = 6.1 Hz,12H).MS(ESI):m/s713(M+H)。其质谱如图1 所示。
1.2.4 N,N-二(2,6-二异丙基-4-羟甲基苯基)苊烯-1,2-二亚胺溴化镍(Cat1)的制备
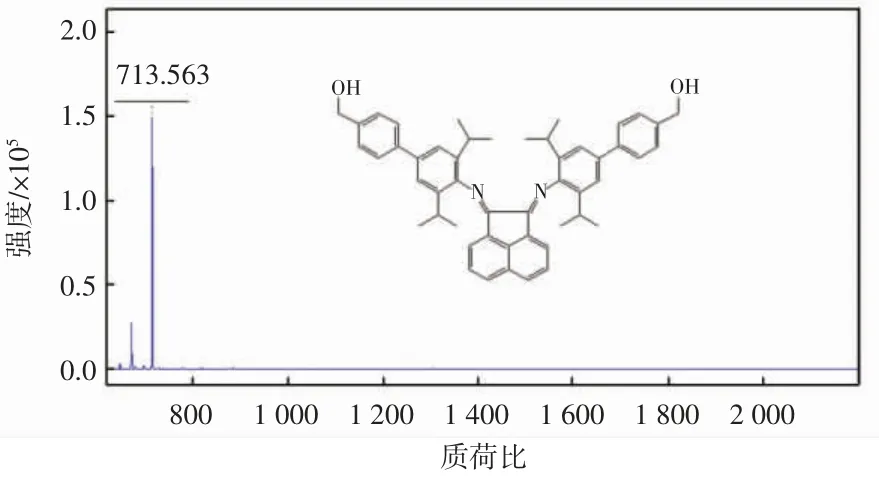
图1 配体L 的质谱图Fig.1 Mass spectrum of ligand L
所有操作均在氩气保护条件下进行。取100 mL三口瓶,向其中加入Ni(DME)Br2(1 mmol)和干燥好的二氯甲烷(20 mL),取 1 mmol 配体 L 溶于 10 mL 干燥好的二氯甲烷中缓慢滴加到三口瓶中,升温至40 ℃,加热回流10 h,之后除去溶剂,用大量正己烷沉淀,多次洗涤后,得橙红色粉末,真空干燥,得到纯产物,产率为87%。
1.3 乙烯聚合实验
全部实验反应均使用Schlenk 标准技术在氩气气氛保护下进行。取100 mL 反应釜,加入搅拌磁子,用氩气置换气体3 次,充入乙烯气体,设定所需温度,并保持一定压力。搅拌中,通过注射器将一定量的甲苯、催化剂和助催化剂一氯二乙基铝(DEAC)加入其中,保持反应压力开始聚合。达到预定聚合时间后,切断乙烯供给,泄压降温,加入10%酸化乙醇终止反应。产物经水和乙醇反复洗涤后抽滤,在60 ℃下真空干燥至恒重,称量并计算其活性:
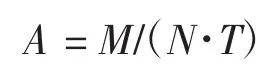
式中:A 为催化剂活性(g/(mol·h));M 为聚乙烯质量(g);N 为催化剂用量(mol);T 为聚合时间(h)。
1.4 配体、催化剂及聚合物表征
1.4.1 配体及催化剂的表征
对配体用AVANGE-400M 型核磁共振波谱仪进行核磁谱图分析,在室温下测试,以氘代氯仿为溶剂;利用Nicolet-iS50 型傅里叶红外光谱仪表征结构,采用 KBr 压片,扫描范围为 4 000~400 cm-1;利用 Varian 7.0T FTMS 型质谱仪对配体进行表征。
1.4.2 聚合物的表征
对聚合物的熔点用DSC200F3 型差示扫描量热仪测定,氮气氛围下进行,温度范围为10~150 ℃,升温和降温速率为10 ℃/min,取第2 次扫描结果,得到熔点Tm,通过熔点的变化来对聚合物支化度进行分析。
2 结果与讨论
2.1 配体及催化剂合成
配体及催化剂的具体合成路线如图2 所示。

图2 配体L 和配合物Cat1 的合成路线Fig.2 Syntheses of ligand L and complex Cat1
由于金属镍的引入,配合物Cat1 表现出的顺磁性使核磁共振难以表征出理想的图谱,因此取用红外吸收光谱对配体和配合物进行表征分析,结果如图3所示。
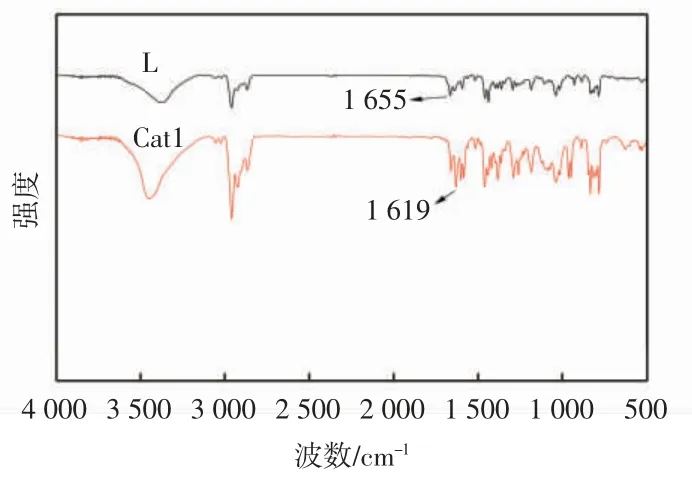
图3 配体L 和配合物Cat1 的红外谱图Fig.3 FT-IR spectra of ligand L and complex Cat1
由图3 可知,配体在波长1 655 cm-1处有明显的伸缩振动峰,此峰为C=N 特征吸收峰。配合物的C=N特征吸收峰位于波长1 619 cm-1处,较配体明显向低频方向移动,证明配合物Cat1 中的N 和Ni 成功配位。
2.2 催化乙烯聚合性能研究
催化乙烯聚合时,以配合物Cat1 为主催化剂,以一氯二乙基铝(DEAC)为助催化剂,在常压或高压条件下进行聚合反应。通过对聚合反应温度、铝镍比(nAl/nNi)的调控,来分析配合物Cat1 对乙烯聚合性能的影响,其结果如表1 所示。
由表1 可知,当聚合压力保持0.5 MPa、铝镍比固定不变、聚合时间相同时,催化剂Cat1 催化乙烯聚合的活性伴随着温度从30 ℃升高到80 ℃呈现出先升高后降低的特点。分析认为,当温度为30 ℃时,温度过低,催化剂配体本身位阻较大,乙烯插入困难。当温度升高至50 ℃时,反应加速进行,催化活性达到最高值4.28×106g/(mol·h)。伴随着温度的继续升高,Cat1 催化活性开始出现下降趋势,表明其部分活性中心开始分解,至80 ℃时,虽然催化活性降低至2.46×106g/(mol·h),但仍然具有较高的应用价值,Cat1 表现出了良好的热稳定性。
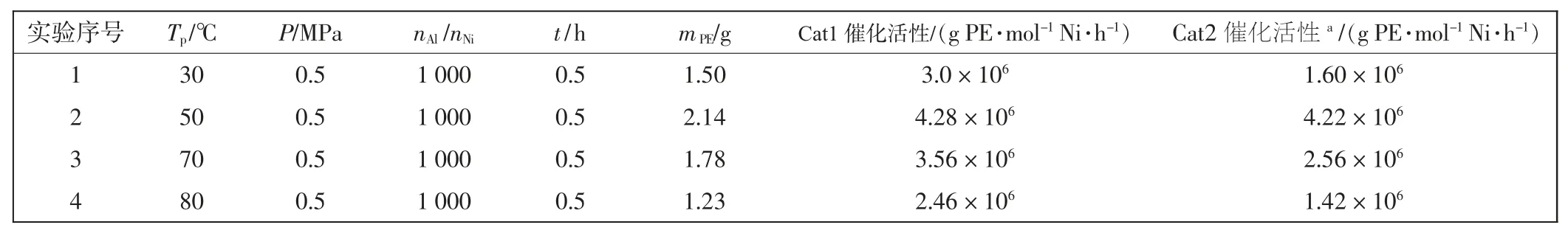
表1 聚合温度对催化性能的影响Tab.1 Effect of polymerization temperature on catalytic propreties
Cat1 与此前报道过的Cat2 为分子质量相同、结构相似的2 种配合物,如图4 所示[17]。但是在相同聚合条件下,这2 种催化剂却表现出差异较大的催化活性。虽然2 种催化剂在50 ℃的聚合温度下表现出的催化活性峰值相近,但是随着温度提高至70 ℃和80 ℃时,Cat2 的催化活性降幅非常明显,而催化剂Cat1 显示出更好的耐高温活性。通过对2 种配合物结构进行对比可以发现,虽然这2 种催化剂具有相同的分子量和相似的化学结构,但是在Ni 原子催化活性中心周围,Cat1 具有更大的空间位阻,较大的空间位阻对活性中心形成保护,降低了活性中心对温度的敏感性,减缓了活性中心在高温下分解的速率,使配合物Cat1 比配合物Cat2 在较高温度下表现出更好的热稳定性。
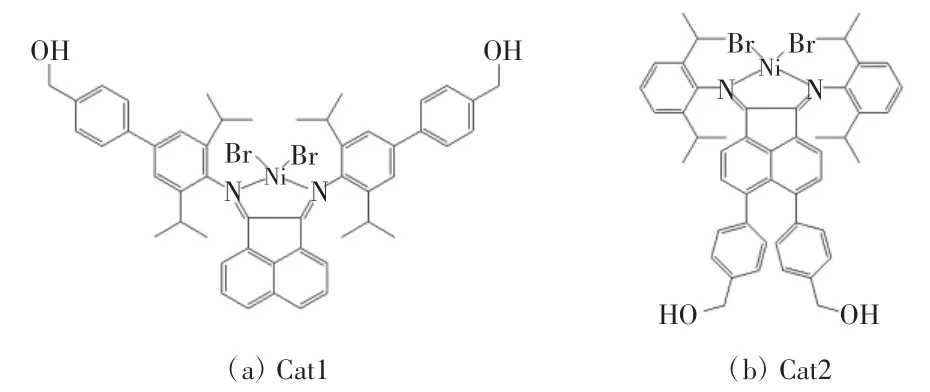
图4 配合物1 和配合物2 的结构对比Fig 4 Comparison of structures of complex 1 and complex 2
表2 为算法与适应度参数值对照表。
由表2 可知,当聚合温度保持为50 ℃、聚合压力不变、聚合时间相同时,通过改变铝镍比(nAl/nNi)来分析催化活性的变化,伴随着铝镍比(nAl/nNi)的不断提高,催化活性呈持续增长的趋势。分析认为,当铝镍比(nAl/nNi)较低时,会使单烷基化反应优先发生于部分助催化剂(DEAC)与主催化剂之间,单烷基化的主催化剂会因被助催化剂夺去电子而成为缺电子阳离子活性中心,使乙烯单体更容易插入,伴随着助催化剂用量的提高,活性中心的形成趋于充足,催化活性逐渐提高。当铝镍比(nAl/nNi)为1 200 时,催化活性达到了较高的4.80×106g/(mol·h),因此,想要达到理想的催化效果,必须严格控制助催化剂的用量。
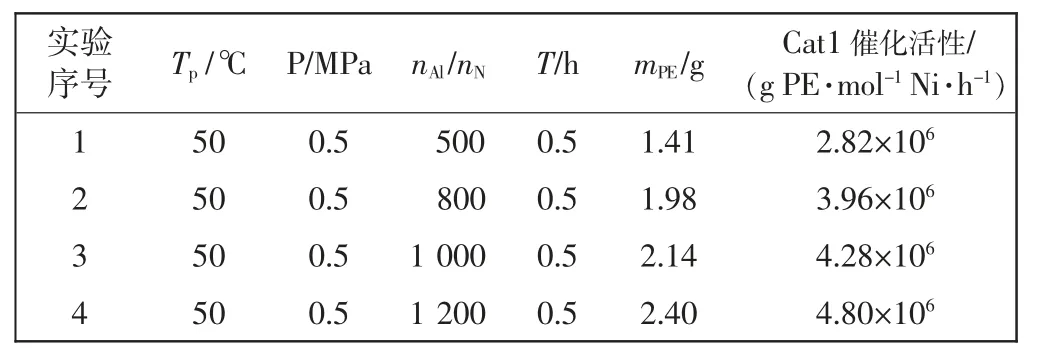
表2 算法与适应度参数值对照表Tab.2 Comparative fitness parameters of methods
2.3 聚合物的表征
对配合物Cat1 催化所得聚乙烯产物进行了13CNMR表征,如图5 所示,通过相关计算,其支链分布结果如表3 所示。
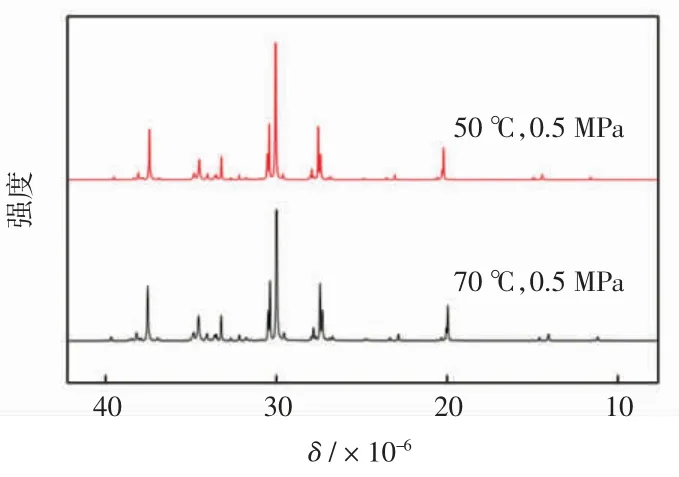
图5 聚乙烯13C NMR 谱图Fig.5 13C NMR spectra of PE
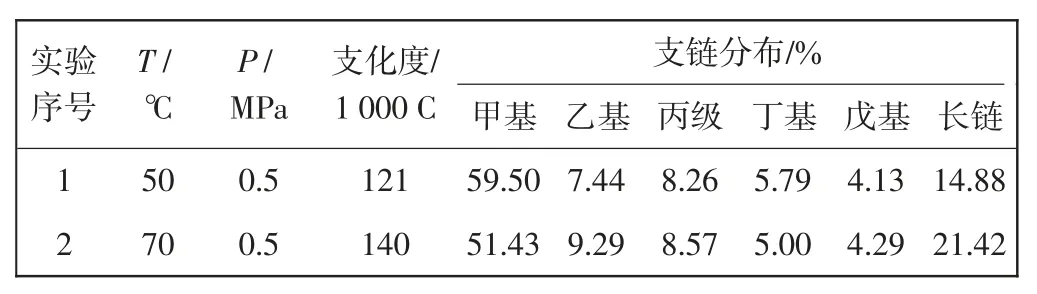
表3 聚乙烯的支化度分布Tab.3 Branching degree of selected polyethylene
由图5 可以明显看出,所得聚乙烯支链结构多样且复杂,不仅包括甲基、乙基等短支链结构,同时含有大量长支链结构。由表3 分析可知,当聚合温度升高时,支链分布发生明显改变,短支链甲基占比由59.50%下降至51.43%,长支链占比大幅上升,由14.88%上升至21.42%。分析认为,伴随着聚合温度的提升,β-H 的消除和乙烯插入速率增大,催化剂在聚乙烯分子链上行走加剧,使聚合物支化度得到提高。
为进一步分析聚乙烯支化度,本文对其进行了DSC 表征,如图6 所示。

图6 聚乙烯DSC 熔点曲线Fig.6 DSC melting curves of PE
由图6 可知,聚合压力均为0.5 MPa,聚合温度为50 ℃时所得聚乙烯熔点为38 ℃,观察聚合温度为70 ℃时所得聚乙烯,未能检测到熔点值。分析认为,随着聚合温度的上升,聚合物支化度提高,产生了更多的支链结构,导致结晶能力变差,与13C NMR 表征结果相一致。
3 结 论
本文通过引入大空间位阻取代基合成了一种新型α-二亚胺配合物,并通过核磁共振、红外元素分析等手段对该配合物进行了表征,通过改变聚合中的反应条件来探索了新合成的配合物在乙烯聚合反应中的催化效果。
(1)该种新型催化剂在80 ℃时仍保持较高的活性,即高活性温度范围变宽。
(2)聚合反应中催化活性与助催化剂的含量密切相关,当铝镍比(nAl/nNi)达到1 200 时,催化活性高达4.80×106g/(mol·h)。
(3)聚合物的性能与聚合温度密切相关,相同的聚合压力下,随着聚合温度的提高,聚合物的支化度相应提高。
