常压密闭微波消解-电感耦合等离子体发射光谱法测定锑矿石中的锑
2020-03-25郑智慷曾江萍王家松乔赵育刘义博吴良英王力强
郑智慷,曾江萍,3 ,王家松 *,乔赵育,刘义博,吴良英,王力强
(1.中国地质调查局天津地质调查中心,天津 300170;2.华北地质科技创新中心,天津 300170;3.中国地质调查局泥质海岸带地质环境重点实验室,天津 300170;4.中检(天津)检测有限公司,天津 300300)
锑是一种不可再生的战略性矿产资源,主要用于阻燃剂、铅酸蓄电池、化学制品及玻璃陶瓷等领域。随着经济发展的需要,我国对锑资源的需求量逐渐提升[1]。因此,锑资源的开发是野外地质找矿工作的热点,准确测定锑矿石中锑元素的含量是锑矿石化学分析的一项重要工作。
常用的样品前处理方法有酸溶和碱熔。李皓等[2]采用过氧化钠碱熔方式对锑矿石进行前处理。该方法可有效避免由于锑含量较高、样品成分较复杂造成的溶样不彻底的问题。但由于碱熔会引入较多盐类,导致样品溶液盐类浓度较高,易造成雾化器堵塞,因此在实际工作中常采用酸溶作为锑矿石的前处理方法。自然界中的锑大多以三硫化二锑的形式存在[3],而锑的硫化物易被王水分解,故目前普遍采用王水消解作为锑矿石的溶矿方法。然而在实际工作中发现,王水消解所得锑的分析结果往往较认定值偏低。这主要是因为岩石中的硅酸盐含量较高,锑的地球化学特征导致锑能够以类质同象形式进入造岩矿物,使部分锑被硅酸盐包裹[4]。由于王水无法溶解硅酸盐,导致硅酸盐中包裹的锑难以被溶解,造成分析结果偏低。魏轶等[5]采用氢氟酸、硝酸、高氯酸、硫酸混合酸溶样作为锑矿石前处理方法。该方法通过引入氢氟酸,较完全地分解样品中的硅酸盐,从而较为充分地释放样品中的锑,避免了锑矿石溶样不彻底的问题。
锑元素的测定方法主要有硫酸铈容量法[6-7]、分光光度法[8]、原子吸收光谱法[9-12]、原子荧光光谱法[13-16]、X射线荧光光谱法[17-21]、电感耦合等离子体发射光谱法(ICP-OES)[22-29]等。其中,容量法和极谱法由于操作过于繁琐,操作难度较高,目前已较少使用;原子吸收光谱法由于线性范围低,已很少使用。锑矿石的边界品位为0.7%,若采用原子荧光光谱法测定,则存在较大的稀释误差。ICP-OES是近些年高度发展的分析测试技术,依靠其高效稳定、可连续快速多元素测定、线性范围宽、精确度高的优点,已广泛应用于岩石、土壤、水系、沉积物中金属、非金属元素的分析测定。目前,应用ICP-OES进行锑矿石的分析虽提高了分析效率,但前处理方式仍较多采用王水溶矿,对锑的溶解不彻底。且ICP-OES多用于较低含量锑的测定,对锑矿石中含量较高的锑(10%~39.7%),由于其相较于低含量锑在溶矿过程中更难充分溶解,又因目前锑的提取过程多采用盐酸介质,难以充分抑制较高浓度锑的水解,高浓度锑在复溶过程中更易发生水解,在实际操作中更易造成测定结果偏低,故分析测定方法相对较少。因此,溶矿过程更为彻底、最大限度避免锑的水解,对准确测定锑矿石中锑的含量具有重要的意义。
本文以锑矿石标准物质为分析对象,采用氢氟酸、硝酸、盐酸混合酸消解体系,氢氟酸可较完全分解样品中的硅酸盐,充分释放样品中的锑。通过对比敞口酸溶、常压密闭微波消解、高压密闭消解等不同溶样方式,择优选择最佳前处理方式;通过对比不同提取介质下锑的测定结果,尝试探究了不同提取介质对锑元素水解的影响,择优选取最大限度抑制锑水解的介质。通过上述的条件对比实验,为锑矿石中锑的准确测定提供了一种更加准确的方法。
1 实验部分
1.1 仪器和工作参数
MARS6高通量密闭微波消解系统(美国CEM公司)。
Optima 8300电感耦合等离子体发射光谱仪(美国PerkinElmer公司)。仪器工作参数为:射频功率1300W,等离子体气流量15L/min,辅助气流量1.0L/min,雾化气流量0.70L/min,泵进样量1.50mL/min,读数延迟时间30s。
1.2 标准溶液和主要试剂
锑标准溶液:移取25mL锑标准储备液(1000mg/L)于250mL容量瓶中,用盐酸-酒石酸混合溶液(5∶95)定容,混匀,得到锑标准使用液(100mg/L)。
氢氟酸、盐酸、硝酸:微电子级,购自北京兴青红精细化学品科技有限公司。
高氯酸:优级纯,购自天津市鑫源化工有限公司。
酒石酸:分析纯,购自天津市科密欧化学试剂有限公司。
1.3 样品前处理
为确定本方法的线性范围,本文选择锑含量各不相同的锑矿石国家标准物质GBW07175(Sb认定值18.97%)、GBW07176(Sb认定值39.7%)、GBW07279(Sb认定值6.26%)、GBW07280(Sb认定值1.81%)进行实验。称取0.0500~0.5000g样品于微波消解罐中。用少量去离子水湿润后,于通风橱中缓慢加入3mL盐酸、1mL硝酸、4mL氢氟酸混匀。若反应剧烈,待初步反应过后再旋紧消解罐。然后按照表1所列的升温程序于微波消解仪中消解,待运行结束后冷却。
表1微波消解升温程序
Table 1 Program of microwave digestion

步骤升温时间(min)目标温度(℃)保持时间(min)功率(W)1510001200251203120035130251200
消解完毕后,取出消解罐,用去离子水将消解液转移至聚四氟乙烯烧杯中,加入1mL高氯酸后,于电热板上加热蒸发。待冒较浓白烟时,用5mL盐酸溶解烧杯内溶物,用5%酒石酸和5%盐酸混合溶液定容,摇匀,待测。
1.4 标准曲线
将锑标准使用液逐级稀释配制成锑标准系列。分别移取1、5、10、20、50、80mL锑标准溶液于100mL容量瓶中,用5%酒石酸和5%盐酸混合溶液定容,摇匀,得到浓度为1、5、10、20、50、80μg/mL的锑标准系列。该标准系列溶液现用现配。采用电感耦合等离子体发射光谱仪对空白及标准溶液进行测定。以标准系列的光谱强度(扣除空白)为纵坐标,锑的质量浓度为横坐标,绘制标准曲线,标准曲线方程为y=554.39x-36.417(R2=0.9999)。
表2不同提取介质的测定结果
Table 2 Analytical results of Sb in sample pretreated with different volumetric methods

标准物质编号Sb认定值(%)定容方式1(5%酒石酸与5%盐酸混合溶液)定容方式2(15%王水定容)定容方式3(20%盐酸定容)4次测定值(%)平均值(%)4次测定值(%)平均值(%)4次测定值(%)平均值(%)GBW0717518.9719.01 18.9919.04 19.0019.0116.24 16.3116.13 16.2016.2218.64 18.5718.69 18.6218.63GBW0717639.739.74 39.8139.77 39.7339.7633.57 33.4933.26 33.3833.4339.36 39.3139.24 39.3839.32GBW072796.266.28 6.316.27 6.296.295.31 5.465.37 5.345.375.97 5.895.91 5.845.90GBW072801.811.83 1.841.80 1.821.821.44 1.511.55 1.531.511.64 1.591.61 1.631.62
2 结果与讨论
2.1 提取介质的选择
对锑矿石标准物质GBW07175、GBW07176、GBW07279、GBW07280分别取4份,按照1.3节的样品前处理方法进行处理,分别采用5%酒石酸与5%盐酸混合溶液[2]、15%王水[23]、20%盐酸[5]作为提取介质,定容后于ICP-OES仪器上进行测定。
不同提取介质对应的分析结果见表2。由表2可以看出,5%酒石酸和5%盐酸介质下(方式1),测得锑的结果最为准确,而其他两种提取介质下锑的测定结果均存在不同程度的偏低。对于这种现象,本文认为,在样品消解的过程中,锑被氧化为五价。五氯化锑在溶液中极易发生水解。王水介质[23]和盐酸介质[5]中,溶液中的H+无法充分地抑制锑的水解,从而造成了测定结果的偏低。而相较于魏灵巧等[23]的分析方法,本文采用5%酒石酸和5%盐酸混合溶液的介质,锑与酒石酸[30]络合,导致锑的水解平衡逆向移动。盐酸的存在,保证了溶液的酸性,从而最大程度地避免了锑的水解,使测定结果更接近认定值。
在实际实验过程中发现,15%王水[23]介质下,溶液中析出的白色沉淀较多;20%盐酸[5]介质中,溶液较为稳定,随着时间的推移仅出现少量沉淀。基于盐酸介质中的沉淀过少,不易获取且不易于X射线衍射仪分析,故只取15%王水介质中的沉淀,经过过滤、洗涤、自然晾干后,于X射线衍射仪上进行测量,得到沉淀的衍射图谱。
从X射线衍射图谱(图1)的特征峰可确定洗涤干燥后得到的沉淀为五氧化二锑,由此可知王水介质中析出的沉淀确为锑的水解产物。沉淀的析出造成了锑的损失,导致了测定结果的偏低。因此,本方法采取5%酒石酸和5%盐酸混合溶液进行提取。

图1 溶样后析出沉淀的X射线衍射图谱
2.2 溶样方式的选择
常用的溶样方式主要有敞口酸溶[22]、常压密闭微波消解[15]、高压密闭消解[24]等。本实验对标准物质GBW07175分别采取氢氟酸、硝酸、盐酸、高氯酸混合酸敞口酸溶、常压密闭微波消解、高压密闭消解三种方式进行消解,所得消解液按照1.3节步骤处理后于ICP-OES仪器上测定锑含量。不同溶样方式的消解时间和条件及测定结果见表3。
表3不同消解方式下锑的测定结果对比
Table 3 Comparison of the analytical results of Sb pretreated with different digestion methods
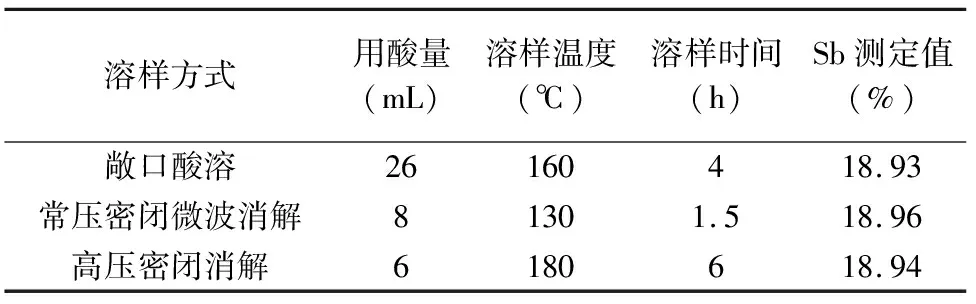
溶样方式用酸量(mL)溶样温度(℃)溶样时间(h)Sb测定值(%)敞口酸溶26160418.93常压密闭微波消解81301.518.96高压密闭消解6180618.94
由实验结果可知,三种消解方式所得测定结果大致相同,均可满足锑矿石中锑含量测定的分析要求。其中常压敞口消解[22]的方式,用酸量最多,对环境的影响相对较大,且溶样时间较长;高压密闭消解的方式,用酸少,但溶样时间过长;常压密闭微波消解的方式,用酸量较少,对环境影响相对较小,且溶样温度低、溶样时间短,避免了较高温度下五氯化锑挥发及分解的问题。
由表3可知,相比敞口酸溶和高压密闭消解,常压密闭微波消解技术[31]同时具有样品分解快速、完全,且消解温度相对较低,挥发性元素损失较少,试剂消耗相对较少,操作简单,处理效率高的特点。
综上,本方法采取常压密闭微波消解的溶样方式。
2.3 特征谱线的选择
在发射光谱上,锑元素有206.836nm、217.582nm[25-27]、231.146nm、252.851nm、204.957nm、203.977nm共六条谱线。应用ICP-OES测定元素时,应选择干扰少、背景低、信噪比高的特征谱线[26]。由于Sb 231.146nm受Co 231.160nm干扰,Sb 203.977nm受Se 203.985nm干扰,Sb 252.851nm受干扰严重,Sb 217.582nm及Sb 204.957nm背景更高且在20μg/mL上的信号强度较Sb 206.836nm分别低1000及10000。相比之下,Sb 206.836nm分析谱线受到的干扰极少,背景较低,信号强度相对较高。综上考虑,本方法选择Sb 206.836nm的特征谱线。
2.4 方法技术指标
2.4.1检出限
用相同的样品处理方法和仪器测量条件,于ICP-OES平行测定空白溶液10次,根据10次空白测定结果,得到标准偏差为0.37%,以3倍标准偏差除以标准曲线的斜率计算得到检出限为1.10μg/g。本文方法检出限与严慧等[22]的检出限(1.58μg/g)大致相同,显著低于李皓等[2]的检出限(50.0μg/g)和魏轶等[5]的检出限(30.0μg/g),略低于魏灵巧等[23]的检出限(5.0μg/g)。本文认为,采取氢氟酸等混合酸溶矿的方式,所得溶液基体类似,均较充分溶解了锑矿石。但所得溶液酸度不同,对仪器性能的影响可能不同,较高的酸度会影响仪器的雾化效率,故魏轶等[5]的检出限较高;魏灵巧等[23]采取王水溶矿,对锑矿石溶解不彻底,且所得溶液酸度较高,可能对检出限造成了一定影响;相比于严慧等[22]的稀王水介质,本文采取了酒石酸-盐酸介质,酸度较低,且酒石酸对锑的络合更有效地抑制了锑的水解,因此检出限更低。不同方法所用仪器不同,仪器性能对锑的检出限可能有不同程度的影响。李皓等[2]的碱熔方法,溶液中盐类较高,基体较为复杂,故检出限高。
表4方法准确度和精密度
Table 4 Accuracy and precision tests of the method

标准物质编号Sb含量(%)分次测定值平均值认定值相对误差(%)RSD(%)GBW0717519.0119.0418.9819.0118.970.210.1119.0018.9919.02GBW0717639.8139.7439.7439.7939.70.230.1139.8239.7739.80GBW072796.316.296.246.296.260.480.536.286.306.34GBW072801.771.731.761.741.810.131.111.741.721.73
2.4.2准确度和精密度
对国家标准物质GBW07175、GBW07176、GBW07279、GBW07280,按照1.3节的处理过程,分别测定6份平行样品,考察该方法的准确度和精密度,所得结果见表4。根据测量所得数据计算得知,该方法的相对误差为0.13%~0.48%,相对标准偏差(RSD)为0.11%~1.11%。该方法结果与李皓等[2]碱熔结果相近,优于严慧等[22]四酸溶矿所得结果。究其原因,本文认为碱熔避免了复杂的矿石组成成分对溶样过程的影响,可较完全溶解锑矿石,且碱熔方法在复溶中采用了酒石酸介质,很好地抑制了锑的水解,故所得结果较好。但碱熔实验过程较为繁琐,故实际生产中采用酸溶方式更为简便。而四酸溶矿复溶过程采用稀王水介质,锑在溶液中出现了不同程度的水解,从而造成准确度和精密度的下降。对GBW07280,由于受仪器工作时正常波动的影响,导致测量结果平均值略低于认定值,但分析方法准确度符合行业标准DZ/T 0130—2006《地质矿产实验室测试质量管理规范》的要求。由准确度和精密度的测定结果可知,该方法准确度较高,且所得结果稳定。
2.4.3加标回收率
湖南是我国锑资源储量最高的省份,勘探程度较高。湖南锡矿山是全球唯一一处超大型锑矿床[32],其中不同含量锑矿石分布相对广泛。对采自湖南锡矿山的锑矿石(大致含量在5%~40%),经过碎样工序,制成粒度≥74μm的样品,分别加入不同浓度的锑标准溶液,按照1.3节的前处理方法进行处理。分别测定未加入锑标准溶液的三个锑矿石样品中锑的含量和这三份加入不同锑标准溶液的样品中锑的浓度,根据测量结果,计算该方法的加标回收率为99.6%~102.0%(表5)。
表5方法加标回收率
Table 5 Spiked recovery of the method

项目Sb测定值(%)样品1样品2样品3称样量(g)0.10000.10000.1000溶液体积(mL)100100100加标前样品溶液测定浓度(μg/mL)8.7415.3132.60加标前样品溶液锑含量(μg)87415313260锑标准溶液浓度(μg/mL)100100100加标体积(mL)102040加标量(μg)100020004000加标后样品溶液测定浓度(μg/mL)18.9135.2272.89加标后样品溶液锑含量(μg)189135227289加标回收率(%)102.099.6101.0
3 结论
本文采用常压密闭微波消解的方式快速处理样品,通过引入氢氟酸充分溶解锑矿石,在复溶过程中采用酒石酸-盐酸介质,以最大限度地抑制锑的水解,建立了氢氟酸-硝酸-盐酸微波消解,高氯酸蒸发赶酸,酒石酸-盐酸提取定容,ICP-OES测定锑矿石中锑的分析方法。本方法较之传统方法,用酸量相对较少,对环境影响程度小,分析时间较短、效率更高,能够快速、准确地测定锑矿石中锑元素含量。ICP-OES具备连续快速测定多元素的优势,根据这一优势,本文通过后续实验条件的优化,可同时分析锑矿石及其他矿石中的伴生元素及常量元素。
