HMX相变与热分解的模拟研究
2020-03-24李命遥
摘要:当前,对HMX的理论研究主要集中在分子动力学模拟方面。利用分子动力学技术可以模拟晶体结构,计算附着能、化学键、振动能谱、能带结构、熵焓吉布斯能等信息,为分析HMX的相变行为和解释HMX的安定性提供理论支撑,并能有效预测晶体生长过程中的结晶方式和热分解过程中的分解方式。本文即对这方面的研究进展进行综述,重点总结了HMX的相变过程和热分解,并展望了HMX的未来发展。
关键词:分子动力学;HMX;相变;分解
1 绪论
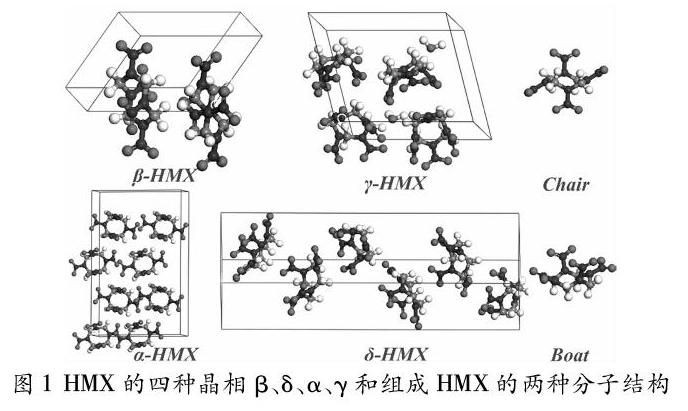
奥克托今(octahydro1,3,5,7tetranitro1,3,5,7tetrazocine)是一種典型的含能材料,综合性能十分优异应用范围极广,代号HMX。HMX晶体结构主要有四种,分别构成β、γ、α、δ相,组成这四种晶相的分子有两种,分别称为船式构象和椅式构象。βHMX的分子结构为椅式,γ、α、δHMX的分子结构为船式。常温条件下稳定存在的相为βHMX,单斜结构空间群为P21/c;γHMX属于混合液态晶相,每个晶胞中γHMX分子和水分子以2:1的形式存在,具有瞬时性和流动性,因此难以通过实验进行γ相的相关探究;αHMX稳定存在的温度范围为103162oC,斜方结构空间群为Fdd2;δHMX在高温状态(160280oC)下稳定存在,六方结构空间群为P61。图1为HMX四种结构的原子构型和两种分子形式。βHMX的应用最广泛,密度为1.89~1.91g/cm,爆轰速度超过9110m/s,爆轰压力超过39.5GPa,能量密度高于TATB,TNT,FOX7等,因此广泛应用于航空航天和国防科技领域。然而,由于HMX存在四种性质不同的晶相,在不同的环境下会发生相变行为,且βHMX的撞击感度较高,因此限制了HMX的进一步发展。从90年代开始,HMX的相变研究已经从单纯的相结构研究转变到载荷条件下的相变研究,从实验研究转变为实验结合理论模拟研究。HMX的相变行为表现出对压强、辐照和温度的高敏感性及复杂多变性,随着计算机技术的发展和量子力学理论的日臻完善,模拟计算能很好的辅助研究HMX在不同环境下的相变行为,并有利于从微观角度和分子尺度上探究HMX的分解行为。HMX的爆炸过程极其复杂,当HMX达到起爆条件时,连锁反应瞬时完成,分子内储存的能量随着小分子(CO,N2,H2O,CO2)的形成与释放,转化成物质波向四周扩散。利用重头计算分子动力学(ab initio molecular dynamics,AIMD)对起爆阶段进行模拟,可以更加直观地认识爆轰过程的能量与物质变化。经典分子动力学模拟(Molecule Dynamics,MD)是一种研究HMX相变的可靠手段,从头计算分子动力学将密度泛函理论应用到分子动力学模拟中,计算结果往往比经典力场分子动力学模拟更加精确,这两种分子动力学方法已经广泛应用于HMX的理论研究中,包括相变研究和热分解研究等。本文即对这方面的研究工作进行综述。
2 分子动力学
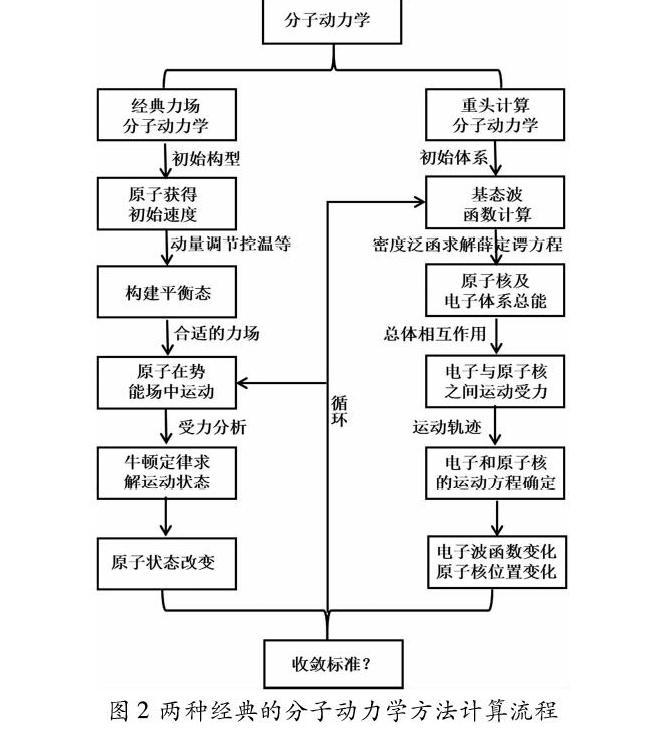
分子动力学是一种基于统计力学采样的分子模拟方法,常用该法研究平衡或非平衡系统的微观细节。对材料开展模拟研究最重要的是结构的能量计算,确保能量最低、模型最稳定,以此提高统计分析结果的可信度。其中分子力学和量子力学则是两种计算能量的方法,分子力学的核心思想是把构成分子的原子当成是弹簧连成的小球,一般称为经典分子动力学模拟;量子力学的核心思想是把原子看成是由电子云连接起来的原子核,一般称为重头计算分子动力学。计算时前者运用对势、多体势(力场),后者基于密度的泛函理论(重头算)计算原子之间的相互作用,得出整个体系的能量变化并分析原子的运动状态。图2列出了两种分子动力学的区别和计算流程。最后,根据平衡态下构像的演化,获得体系的晶体结构、键长、结合能、光学性质和力学性质等,通过所得的晶胞参数、键长、键角、键能变化探索材料的结构特点、理化特征和分解过程等。经典分子动力学主要用于研究凝聚态HMX,包括模拟压强和温度对HMX相变的影响,并跟据晶体结构以及热、动力学结果解释相变的发生过程;从头计算分子动力学主要用于HMX的热分解途径及反应机理研究,虽然起爆时是否反生相变这一问题存在很大的争议,但是根据分子结构、能量和键长的变化可以客观的解释不同的反应途径。利用分子动力学方法从分子尺度出发研究HMX的相变、热分解、感度、稳定性,一直以来都是含能材料计算领域里的研究热点和难点。第三部分将从经典分子动力学和重头计算分子动力学两方面概述分子动力学在HMX中的应用。
3 分子动力学模拟
3.1 单质HMX的经典分子动力学模拟
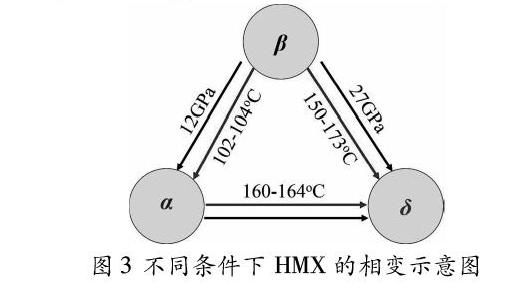
经典分子动力学中最重要的是选择合适的力场势函数。目前,常用CAMPASS(凝聚态优化分子势原子力场)、SB(SmithBharadwaj分子柔性力场)、ReaxFF(反应性力场)等力场来描述含能材料HMX各原子之间的静电相互作用以及分子之间的范德华力。崔红玲[1]等人用CAMPASS力场描述不同温度和不同压强范围内分子间的势能变化,通过动力学优化得出了各相基态HMX的晶胞参数和晶胞体积的变化情况,计算出β、δ、α相的杨氏模量、弹性模量、剪切模量、泊松比等力学参数,佐证了常温加压时β→δ相变在27GPa时出现,常压升温时β→δ相变在440K时出现。Nithin Mathew等根据实验结果修正了SB力场中NO和CH的相互作用,计算出了βHMX在0K、10430GPa等静压条件下的二阶弹性刚度张量和各向弹在模量,结果表明:弹性系数随着压强的增加而单调增加,且绝对温度下在30Gpa内HMX没有相变。周婷婷等[3]用ReaxFFlg力场模拟了不同温度条件下HMX相变导致的体积膨胀效应,将平衡态晶胞参数与温度进行拟合,计算出不同升温过程中的线膨胀系数和体膨胀系数,并通过分析晶胞参数的突变情况确定了相变温度范围。根据分子动力学的研究结果,总结出HMX的相变情况如图3所示。各相稳定存在的温度范围:βHMX为30150℃,δHMX为160280℃和αHMX为103162℃。
3.2 凝聚态HMX的重头计算分子动力学模拟
凝聚态HMX的热分解途径和热分解速率受外界环境和内部结构影响。含能材料受到外界的强烈刺激时内部能量急剧增加,对于局部区域能量突然达到材料的引爆点的情况,研究者提出了“热点”论与“热点模型”。含能材料的内部缺陷也会触发热分解中的化学反应从而致爆。NNO2键断裂在热分解过程中占主导地位,重头计算分子动力学研究指出凝聚态中存在“弗伦克尔缺陷”(分子空位)时,HMX分子的断键行为首先发生在空位周围。晶体内部HMX分子的激活能也比界面和空位缺陷位置上的HMX分子高12kcal/mol,Kuklja等人也指出內部NNO2断裂所需能量比界面和空位缺陷位置高67kcal/mol。分解时缺陷处和界面处的反应速率与气相的反应速率相当,空位浓度越高热分解中的反应速率常数越高,反应活化能越低,因此空位浓度将促进热分解过程的反应。分解温度较低时(1500K)时空位缺陷将促进各种分解途径,较高时(2500K)空位缺陷将阻碍HONO形成。重头计算分子动力学还指出HMX的热分解和孔洞的形状大小有关,不同的孔体积形式对冲击、热辐射等外场作用的响应方式不同,因此热解过程中的断键、成键和能量也存在差异。HMX的撞击感度决定于气泡的空间形状圆柱型、球型、平板型和椭球型等,通常三维孔型比二维孔型对外界诱因更敏感。
不同于块体和粉末炸药的热分解,刘智超[2]等人利用重头计算分子动力学研究了纳米HMX的热分解,对比了不同初始分解路径下分子的解离方式,HMX纳米粒子整体的热分解很大程度上依赖于分解温度和粒子的纳米尺度,其分解机制和分解动力学与块体明显不同。由两个连续主要的步骤控制:一、表面纳米HMX颗粒快速膨胀和单分子分解之间的竞争。二、复杂单分子和双分子竞争分解。另外,低温和高温情况下的分解反应也不相同,低温下HMX发生分子内异构化反应,高温下发生开环氮原子均裂反应。目前,理论研究中很少描述缺陷之间的相互作用,计算时通常忽略它们的影响。实际晶体结构比理想晶体结构缺陷浓度高,因此感度更高。
3.3 气态HMX的重头计算分子动力学模拟
HMX的相变机制和热分解机制不同,气相与凝聚态的热分解机制也存在差异。重头计算分子动力学的研究结果表明,气相分解时通过单分子通道进行,主要的热分解方式包括:HMX分子协同环裂解,NNO2键均裂,硝基硝酸盐重排(CN建断裂),亚硝酸释放等,热分解途径如图4所示。分解途径需要的活化能为3645kcal/mol,但不同的反应机制对应不同的后续分解化合过程,最后以一氧化氮,二氧化碳,水,氮气等小分子形式释放。利用重头计算分子动力学可以探索HMX分子的分解方式,计算HMX分解过程中断键激活能、原子重排活化能、中间产物形成能以及各键长变化,解释起爆时HMX分子的微观结构变化。如果将重头计算分子动力学研究HMX的热分解过程和第一性原理研究HMX晶体结构的电子云、电荷密度变化结合起来,将能更清楚的分析起爆至爆炸各阶段的能量释放情况,进一步解释爆炸性能。
4 展望
高性能计算机的快速发展和日益精进的模拟技术,使得计算模拟效率越来越高,并且随着理论物理在计算中的发展与深入,计算结果的精确度得以提高,其结论也越来越可靠,有利于从微观结构分析实验中的新奇现象并解释相关反应机理。
本文阐述了含能材料奥克托今在理论研究领域中的发展情况。研究者们在HMX的相变和热分解研究中已经取得了显著的成就,但是对相变过程中构像的转变细节却鲜有报道。比如相变转化过程中构像转变的能垒大小以及是否存在一种过渡态,这些科学问题有待进一步论证。应当指出,随着模拟计算的发展:(1)通过分子动力学模拟设计高能、顿感的含能材料已经成为可能。组分设计,含氮量越高能量密度越高;结构设计,笼型分子聚集越密集、相互牵连越强爆轰性越好。炸药分子越稳定,感度越低。(2)通过理论研究分子和晶体结构的演化过程,有利于指导HMX的储存,防止老化,杜绝安全事故的发生。(3)对于爆炸的理论研究主要集中于热分解途径的探索和产物的确定,模拟前提为理想状态,如果进行理论计算时充分考虑晶体缺陷、结构畸变、孔洞等的影响,并将这些实际结构用模型建立,有助于研究HMX真实的爆炸性能。毋庸置疑的是理论计算手段能很好的帮助解释实验现象,并能从原子尺度分析各种性能的表现。
参考文献:
[1]Hong Ling Cui,Guang Fu Ji,Xiang Rong Chen,et al.Phase Transitions and Mechanical Properties of Octahydro1,3,5,7Tetranitro1,3,5,7Tetrazocine in Different Crystal Phases by Molecular Dynamics Simulation[J].Journal of Chemical & Engineering Data,2010,55(9):312129.
[2]Zhi Chao Liu,Wei Hua Zhu,Guang Fu Ji,et al.Decomposition Mechanisms of ΑOctahydro1,3,5,7Tetranitro1,3,5,7Tetrazocine Nanoparticles at High Temperatures[J].The Journal of Physical Chemistry C,2017,121(14):772840.
[3]周婷婷,黄风雷.Hmx不同晶型热膨胀特性及相变的reaxff分子动力学模拟[J].物理学报,2012,61(24):24650101.
*通讯作者:李命遥。
