基于转录组测序探究PM2.5损伤肺上皮细胞BEAS−2B的相关机制
2020-03-20胡玲娟何翔张雷冉琴李国平
胡玲娟,何翔,张雷,冉琴,李国平,△
PM(Particulate Matter)是空气中大小不一的颗粒物混合体,其聚集了各种化学和生物元素,如挥发性有机化合物(VOCs)、多环化合物芳香烃(PAHs)、重金属、细菌和病毒等。近年来,以PM2.5(粒径≤2.5µm)和PM10(粒径≤10µm)为主要污染物形成的雾霾天气持续影响着中国的大部分地区,与肺癌[1]、哮喘[2]等呼吸系统疾病有着密切联系。PM2.5和其他粒径较大的大气颗粒物相比,粒径相对更小,面积更大,活性更强,更容易附着有毒、有害物质(重金属、微生物等),且在大气中的停留时间更长、输送距离更远,易避开气管细胞纤毛等过滤机制进入下呼吸道,严重危害肺脏的健康。大量的流行病学资料和临床研究显示PM2.5的暴露与呼吸系统和心血管系统的发病率和死亡率密切相关。PM2.5对肺与心血管造成的损害有肺功能的损害、慢性咳嗽支气管炎和哮喘发病率的增加、加速动脉粥样硬化的发生发展、静脉血栓的形成[3−5]。
转录组测序是对特定细胞在某一功能状态下所能转录出来的所有RNA的总和进行高通量测序[6]。转录组研究是基因功能及结构研究的基础和出发点,通过新一代高通量测序,能够全面快速地获得某一物种特定组织或器官在某一状态下的几乎所有转录本序列信息,可用于研究基因功能和结构、新转录本预测和可变剪接等,已广泛应用于基础研究、临床诊断和药物研发等领域。本实验通过研究暴露于PM2.5环境下肺上皮细胞BEAS−2B损伤相关机制,分析PM2.5对肺上皮细胞BEAS−2B造成损伤的差异基因表达情况,探究PM2.5可能作用的信号通路。
1 材料与方法
1.1 材料 人正常肺上皮细胞BEAS−2B(上海吉凯基因科技有限公司,中国),RNA simple Total RNA Kit总RNA提取试剂盒(Vazyme,中国),RT SuperMix试剂盒和SYBR Green Realtime PCR Master Mix试剂盒(北京天根生化科技有限公司,中国),DMEM高糖培养液(Hyclone公司,美国),0.25%胰酶(Gibco,加拿大),DMSO(Sigma公司,美国),TRIzol试剂(Invitrogen,美国),PM2.5(北京高能物理研究所核分析技术重点实验室,中国)。SB−100DT超声波清洗机(宁波新芝生物科技股份有限公司,中国),实时荧光定量聚合酶链反应(PCR)仪(Ligh−Cycler480II,德国Roche)。
1.2 方法
1.2.1 细胞培养及分组 BEAS−2B细胞常规传代培养。培养基采用含10%小牛血清、高糖DMEM培养液,于37℃、5%CO2饱和湿度环境的培养箱中贴壁培养,每隔1~2 d换培养液1次,待细胞生长达80%融合时,用含EDTA的胰酶消化细胞,相差显微镜下见细胞收缩,变圆,细胞间隙清晰后加入含10%小牛血清的DMEM培养液,按1.5×106个/皿将细胞接种于10 cm培养皿中,分为对照组和PM2.5组,放置37℃、5%CO2培养箱中培养。将购买的PM2.5用超纯水制成10 g/L的储备液,并经高压灭菌处理。按照参考文献[7]中PM2.5的制备方法,用完全培养液稀释PM2.5储备液,制得浓度为200 mg/L的PM2.5工作液。铺板24 h后吸去细胞培养皿中原有培养基,实验组中加入PM2.5工作液,对照孔中加入等量PBS,每组3个平行孔,对照组为PBS_1、PBS_2、PBS_3,PM2.5组为PM2.5_1、PM2.5_2、PM2.5_3,继续培养24 h[7]。
1.2.2 总RNA提取 将经处理的BEAS−2B细胞用PBS洗涤2~3次,并于−80℃冰箱冻存,用于后续RNA的提取。采用TRIzol法提取总RNA,并使用DnaseI消化DNA,总RNA质量和纯度用琼脂糖凝胶电泳检测,RNA完整性用Agilent 2100检测。检验吸光度A260/280,范围在1.8~2.2;所有样本总RNA完整性:RIN>7.0,28S∶18S>1.0。则RNA质量满足建库要求。提取的RNA于−80℃冰箱保存备用,用于转录组测序送样待检。
1.2.3 转录组测序 RNA文库制备以及测序由上海美吉生物医药科技有限公司完成。cDNA文库的构建方法参照美国Illumina生物技术公司的样品制备试剂盒说明书,文库构建完成后使用RNA分析试剂(Agilent 2000)进行库检,库检合格后利用Illumina HiSeq2500高通量测序平台对文库进行测序,并对原始测序数据进行过滤,得到高质量的Clean Data。随后将6个样品的原始测序数据进行质控,去除接头、剪切末端低质量碱基等,利用TopHat2将质控后的序列(Clean Data)与指定的参考基因组进行比对,获得用于后续转录本组装、表达量计算等分析的mapped data(reads)。利用6大数据库[(Evolutionary genealogy of genes)、GO(Gene Ontology)、KEGG(Kyoto Encyclopedia of Genes and Genomes)]对mapped data(reads)进行基因功能注释。
1.2.4 转录组注释 利用6大数据库(NR、Swiss−prot、Pfam、COG、KEGG、GO)对mapped data(reads)进行基因功能注释。利用RSEM软件进行基因表达定量分析,采用TPM(Transcripts Per Million reads)进行表达量计算,基因差异表达的输出数据为基因表达水平分析中得到的read count数据。
1.2.5 差异表达基因(DEGs)筛选 采用TMM对read count数据进行标准化处理,再利用差异基因分析软件DEGseq2进行差异表达基因进行统计,以P≤0.05且|FC|≥1.2为标准,筛选出差异表达基因。
1.2.6 GO富集及KEGG通路分析 采用goatools软件对差异表达基因进行GO富集分析,并对富集结果进行差异表达基因数量统计。利用KEGG功能富集分析,确定差异表达基因在各信号通路上分布的数量,利用超几何分布计算方式,以P<0.05作为阈值,富集通路并统计差异表达基因数量。
1.2.7 实时荧光定量PCR(qRT−PCR)验证 收集对照组和PM2.5组细胞并提取总RNA,逆转录后进行实时荧光定量PCR实验。使用Primer−BLAST,根据转录组相关基因的序列设计基因特异性引物(表1)。目的基因包括核转录因子激活蛋白−1转录因子亚单位(FOSB、FOSL1)、基质金属肽酶1(MMP1)、集落刺激因子2(CSF2)、白细胞介素−6(IL-6)和黏蛋白5AC(MUC5AC)。将提取的RNA逆转录为cDNA后依据SYBR Green PCR Master Mix试剂盒说明书取2µL cDNA进行实时荧光定量反应。反应条件:95℃5 min;95℃10 s,58℃30 s,40次循环。在实验中以Actin为内参照。用2−ΔΔCt法计算相对表达量,并与RNA−seq测序后相关基因表达量进行比较。

Tab.1 Primer sequences of the tested genes used in the qRT-PCR表1 基因qRT-PCR引物序列
2 结果
2.1 RNA的提取和检测 2组细胞总RNA提取较为完整,A260/280均在1.8~2.2之间,浓度均在500 mg/L以上,通过对总RNA进行质检,RIN均>7.0,符合建立转录组文库的总RNA样品的测序质量要求。见表2。
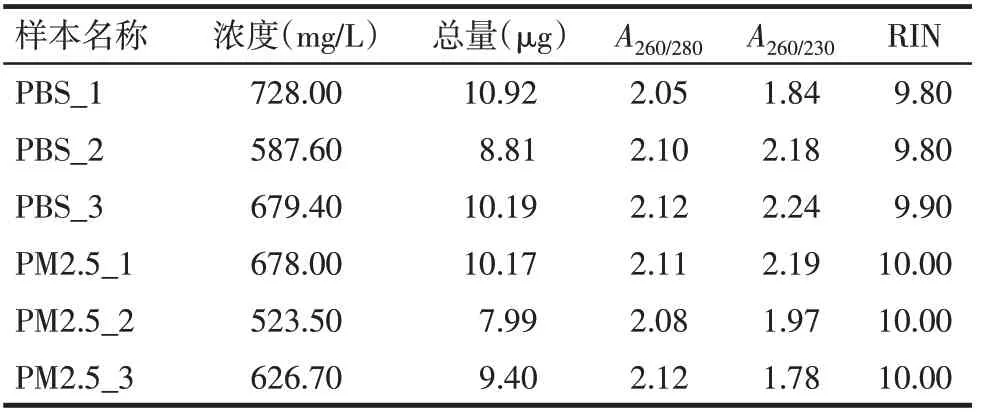
Tab.2 Total RNA quality in transcriptome sequencing samples表2 转录组测序的样本提取的总RNA情况
2.2 转录组测序数据统计 经过测序质控,各样本Clean Data中Q30碱基百分比均≥92.25%,GC碱基所占比例≥51.43%。将Clean Data于参考基因组进行序列比对,从比对结果统计来看,各样本的reads与参考基因组的比对率在97.55%~97.95%。见表3。
2.3 基因的功能注释 6大数据库比对结果显示,有45 044个基因比对到NR数据库,有38 906个基因比对到Swiss−Prot数据库;有16 509个基因比对到Pfam数据库;有39 963个基因比对到COG数据库;有31 469个基因比对到GO数据库;有26 633个基因比对到KEGG数据库,见表4。31 469个基因在GO注释中被分为3大类:生物学过程(biological process)、细胞组分(cellular component)、分子功能(molecular function),共30个功能组(图1)。这些基因主要参与细胞过程(cellular process)、代谢过程(metabolic process)、生理调节(biological regulation)和单有机体过程(single−organism process)等生物学过程。对比到KEGG数据库的基因有26 633个(图2),其中绝大多数基因集中在“生物体系统(organismal systems)和人类疾病”代谢通路中。
2.4 暴露于PM2.5环境中差异表达基因的筛选 共得到961个差异表达基因,其中上调基因有453个,下调基因有508个,差异表达基因的Volcano−plot分布图见图3。采用聚类分析找出具有相同生物学过程或相似功能的基因,结果表明PM2.5处理的肺上皮细胞与未暴露于PM2.5环境的肺上皮细胞样本的生物学过程和功能区分明显,见图4。
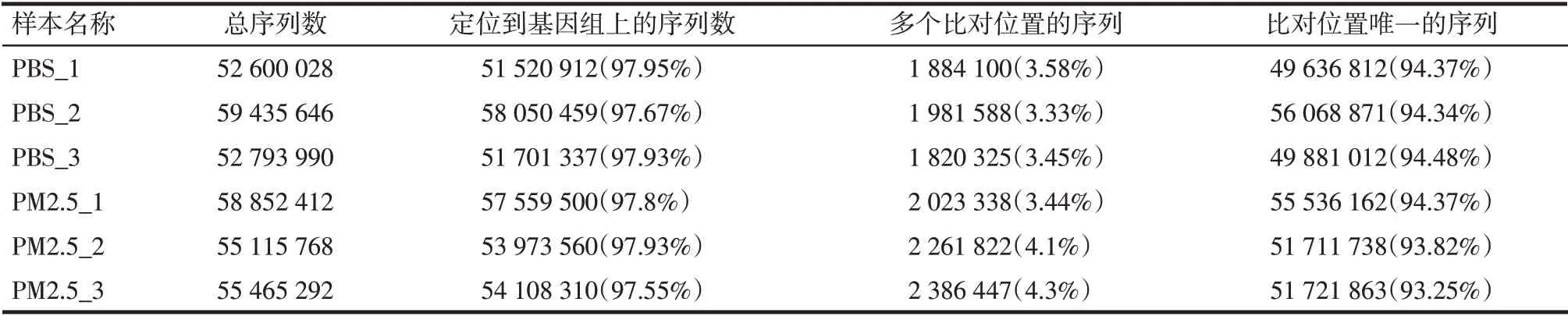
Tab.3 The results of the sequencing sample compared with reference genome表3 测序样本与指定参考基因组的序列比对结果
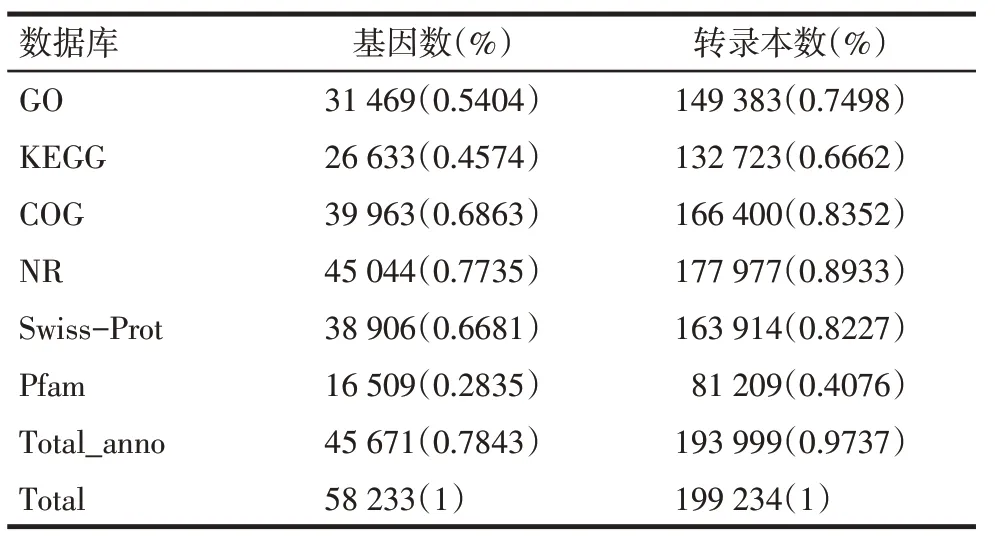
Tab.4 Sequenced genes and transcriptional information in six major databases表4 注释到六大数据库的测序基因和转录本信息
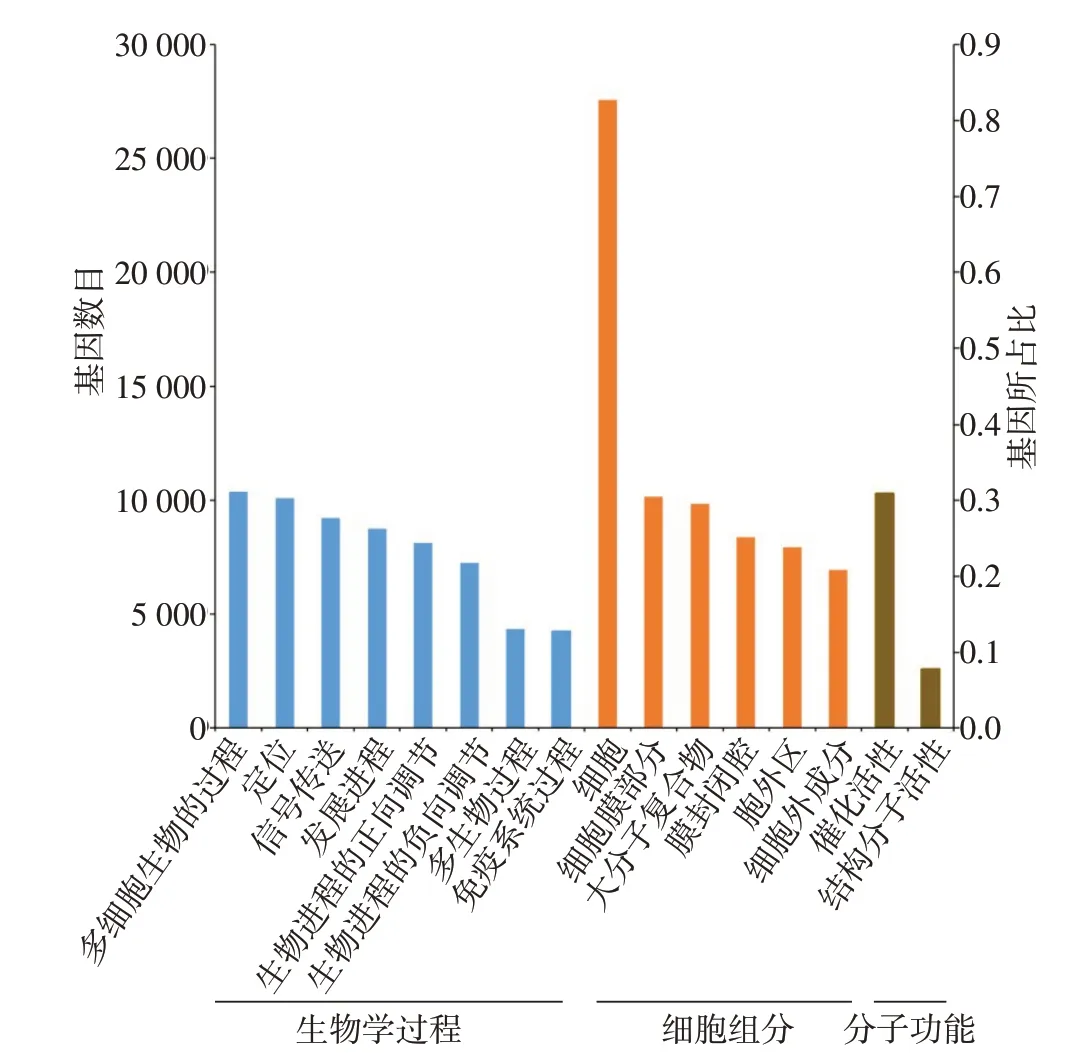
Fig.1 GO classification of genes图1 转录组的GO分类图
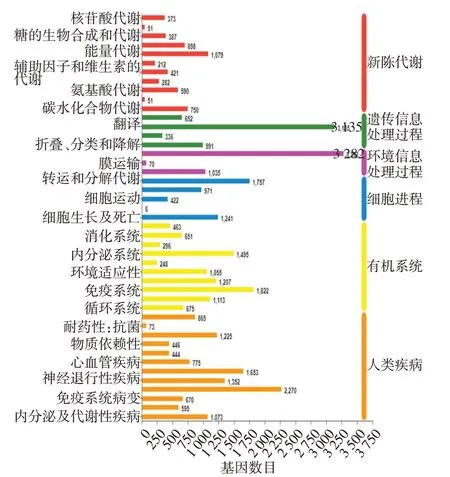
Fig.2 KEGG annotation of genes图2 转录组的KEGG代谢通路途径注释
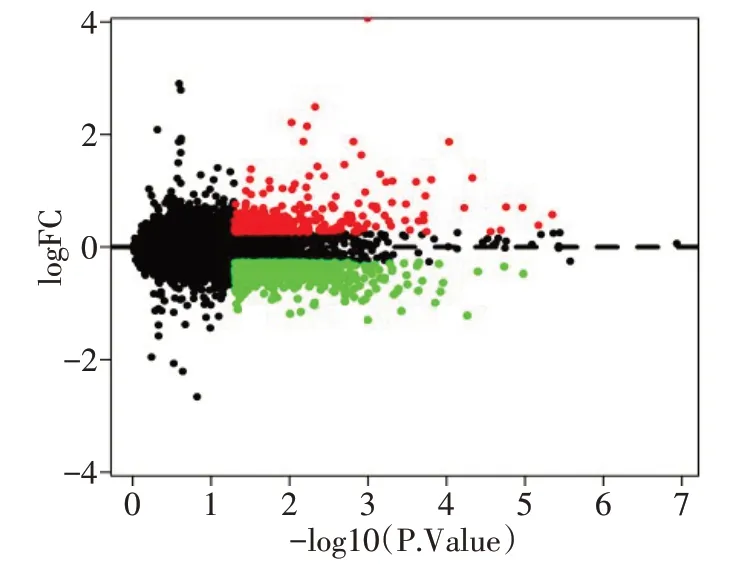
Fig.3 Volcano plot of distribution trends for differentially expressed genes in PM2.5 exposure group and control group图3 PM2.5实验组和对照组差异表达基因的Volcano−plot分布
2.5 差异表达基因GO和KEGG富集分析 对各样本进行聚类分析时,发现PM2.5_2样本中基因与PM2.5_1和PM2.5_3样本中基因表达模式和变化趋势明显不同,出现离群现象,因此在进行后续分析时为保证分析结果的可信度,未将PM2.5_2样本纳入分析。GO分析结果显示,差异表达基因显著富集在2 303个GO terms中,1 812个涉及生物学过程,221个涉及细胞组分,270个涉及分子功能。生物学过程分析(图5A)显示PM2.5参与细胞蛋白质的磷酸化(regulation of protein phosphorylation)、免疫调节过程(regulation of immune system process)、信号转导途径的调节(positive regulation of signal transduction)以及凋亡途径的调控(regulation of apoptotic process)等,并通过调节一系列的蛋白激酶的活动对BEAS−2B细胞蛋白质的代谢、细胞的活动,增殖以及凋亡造成显著影响。从分子功能分析差异基因显著富集在配受体活动(receptor ligand activity)、细胞黏附因子的活动(cell adhesion molecule binding)以及生长因子的活动(growth factor activity)等功能区(图5B)。差异表达基因KEEG富集分析结果显示,差异基因参与机体多种重要的生物学通路(图6),提示PM2.5通过细胞的相互作用、对相关信号通路的调节影响细胞的增殖、迁移以及凋亡等。其中“IL−17信号通路”与细胞增殖、凋亡、代谢以及对病原微生物的免疫应答、中性粒细胞的募集密切相关。富集到此通路的12个差异表达基因包括FOSB、FOSL1、MMP1、MUC5AC、CSF2、TGF-β激活激酶1(TAB3)、IL-6、趋化因子配体(CXCL2、CXCL8),这些基因在PM2.5组的相对表达量均显著上调(P<0.05);而趋化因子配体6(CXCL6)和丝裂原激活蛋白激酶11(MAPK11)、肿瘤坏死因子受体相关因子−4(TRAF4)的相对表达量均显著下调(P<0.05)。见表5。
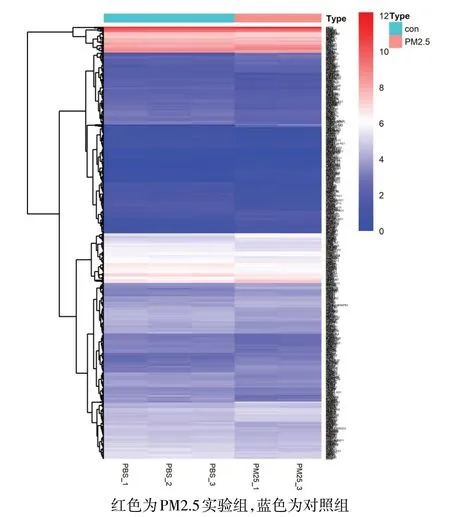
Fig.4 Heat map of differential genes clustering(Red:PM2.5 group;Blue:control group)图4 差异基因聚类热图
2.6 qRT−PCR验证 为了验证PM2.5处理之后差异表达基因富集到“IL−17信号通路”的表达量变化的真实可靠性,本研究选择显著上调的5个差异表达基因(FOSB、FOSL1、MUC5AC、CSF2、IL-6)设计特异性引物后进行荧光定量PCR验证。实验结果表明经PM2.5处理后FOSB、FOSL1和IL-6较对照组表达上调(P<0.05),见图7A,5个关键基因差异表达倍数与RNA−seq结果对应的差异基因表达倍数趋势基本一致,图7B。
3 讨论
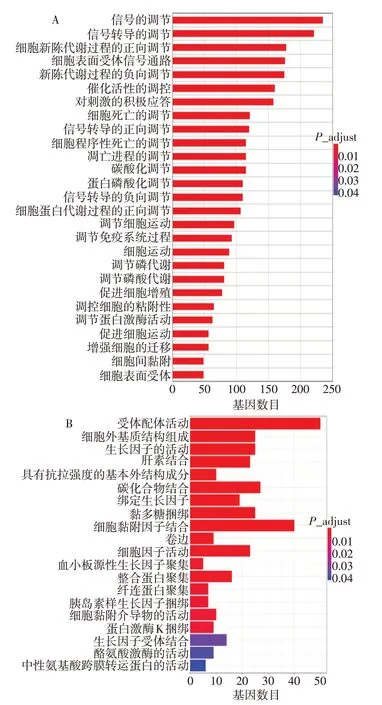
Fig.5 GO enrichment analysis of the differentially expressed genes图5 差异基因的GO富集分析
Th17细胞的分化是由活化的辅助T细胞在TGF-β、IL-6、IL-1β和IL-23存在的情况下所介导的,这些细胞参与了多种自身免疫性和炎性疾病的发展或发病过程,包括肺部疾病,如囊性纤维化、哮喘、慢性阻塞性肺病和肉芽肿病。作为CD4+辅助T细胞的一个独特的亚群,Th17细胞的特点是产生促炎因子IL-17A、IL-17F、IL-22。且IL−17A/IL−17R信号通路通过固有/适应性免疫细胞和结构细胞(如气道上皮细胞AECs)参与了气道免疫系统的形成[8]。而IL-17作为IL-17细胞因子家族的创始成员,可以促进T细胞的激活和刺激上皮细胞、内皮细胞、成纤维细胞产生多种细胞因子如IL−6、IL−8、粒细胞−巨噬细胞刺激因子(GM−CSF)和化学增活素及细胞黏附分子1(cellular adhesion molecule 1,CAM−1),从而导致炎症的产生。在Doe等[9]的研究中发现,IL−17A与受体结合后,可通过MAP激酶途径和核转录因子NF−κB途径从而有效地介导中性粒细胞动员的兴奋过程,并通过刺激骨髓基质细胞产生G−CSF来促进中性粒细胞的募集,而G−CSF又可以促进粒细胞的生成和CD34+细胞内中性粒细胞祖细胞的转化。IL−17A还通过刺激上皮细胞趋化因子配体CXCL1、CXCL2、CXCL5和CXCL8的表达,诱导中性粒细胞募集,介导组织的炎症反应[10]。已有研究证实,IL-17A在哮喘患者的肺、痰标本、支气管肺泡灌洗液(BALF)和血清中的表达增加,这种细胞因子诱导Th2细胞活化、气道高反应、中性粒细胞浸润和黏液生成[11]。而且,哮喘的严重程度也已被证明与IL−17的表达水平呈正相关。此外,由感染引起的肺部炎症(主要是肺炎球菌感染)或自身免疫性疾病(如囊性纤维化)也被认为是由于肺内IL−17a水平的调节所致[12]。可见,IL−17在众多的肺部疾病中发挥着关键作用。另有研究发现,PM2.5后能够沉积于气道和肺上皮,并且缓慢移动到细胞间隙,引起促炎症细胞因子和黏附因子的产生,如IL−1β、IL−6、IL−8,肿瘤坏死因子-α(TNF-α),细胞间黏附因子1(ICAM−1)等。这些促炎症因子的释放会立即启动患者肺部的炎症反应,加重呼吸道症状,尤其是存在心肺基础疾病的患者以及老年人[4,13−15]。虽然,已有研究报道,PM2.5能够激活NF−κB信号通路促进细胞的炎症应答。且PM2.5能激活凋亡相关信号级联,促进细胞凋亡,从而导致肿瘤相关通路失调[16]。但是,PM2.5通过调控IL−17信号影响下游生命进程的研究较少。本研究通过对暴露在PM2.5环境中的肺上皮细胞BEAS−2B进行转录组高通量测序[17−18],探究PM2.5对BEAS−2B细胞造成损害的相关机制,通过对PM2.5处理的BEAS−2B细胞与对照组细胞进行转录组测序,找出暴露于PM2.5环境下的差异表达基因,并对这些基因进行GO功能和KEGG信号通路富集分析,发现这些差异表达基因参与了机体的多种生物学过程,尤其是免疫系统的调节,信号转导途径的调控和细胞增殖与凋亡的调节;KEGG富集分析结果表明“IL−17信号通路”在暴露于PM2.5环境的BEAS2B细胞中起着重要作用,且富集于该通路的基因FOSB、FOSL1、MMP1、MUC5AC、CSF2、IL-6、CXCL2、CXCL8和TAB3的表达量是显著上调的。这表明PM2.5可能通过调控“IL−17信号通路”中关键基因的表达,加重了细胞的炎症反应,促进了细胞凋亡等生命进程。
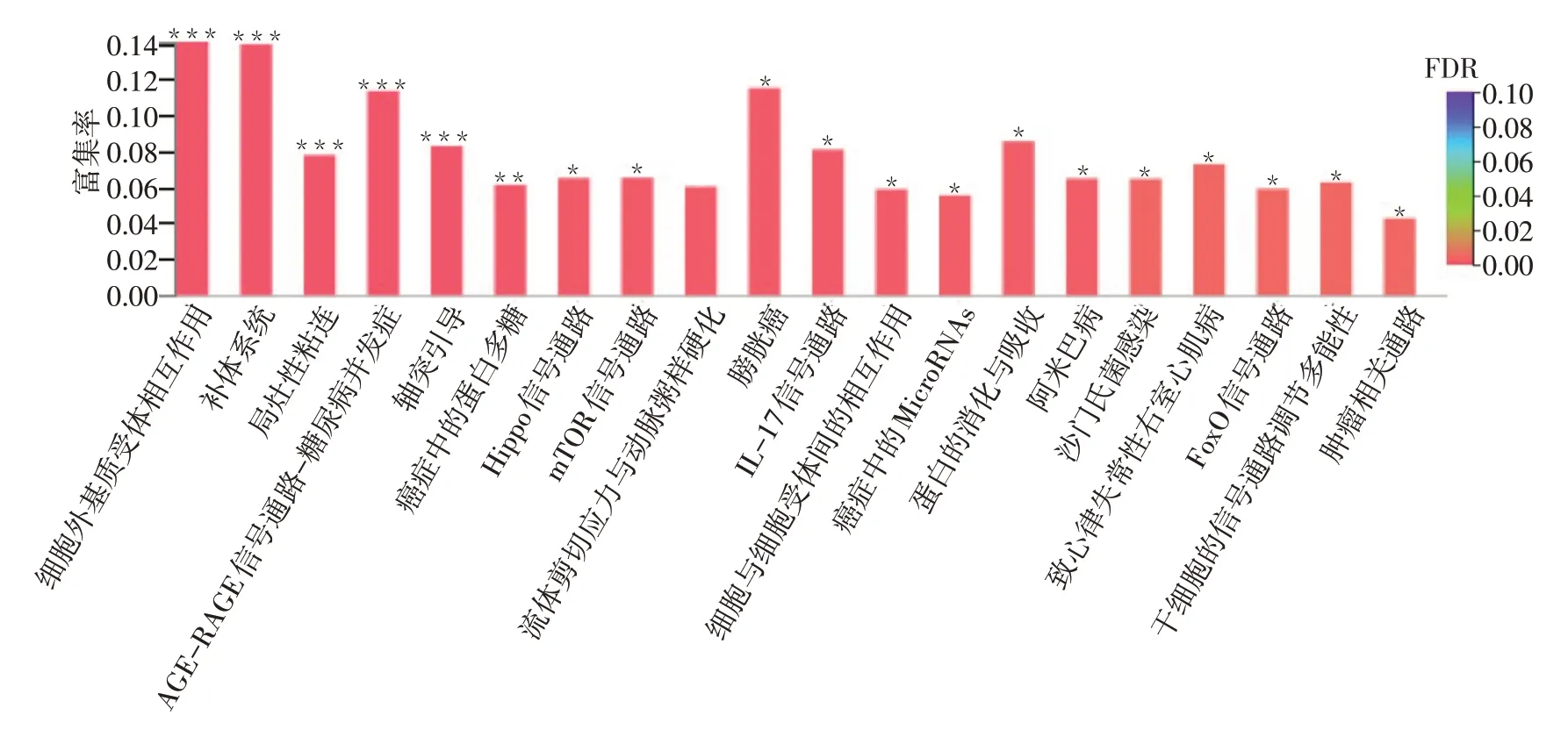
Fig.6 KEGG enrichment analysis of the differentially expressed genes图6 差异基因的KEGG富集分析
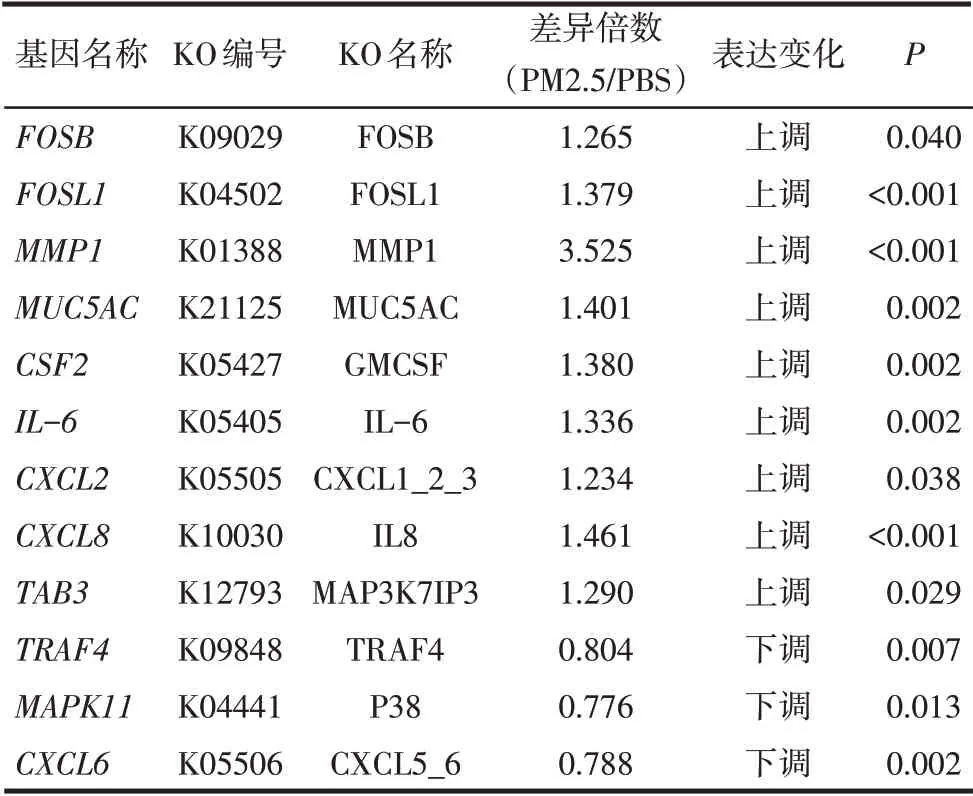
Tab.5 Genes enriched to the"IL-17 signaling pathway"表5 富集到“IL-17信号通路”的基因
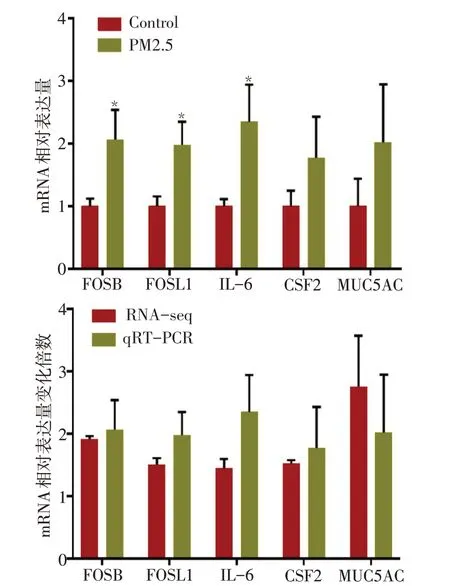
Fig.7 Validation of transcription results with qRT−PCR图7 qRT−PCR验证转录组结果
