散发性克-雅病脑磁共振和脑电图长期随访
2020-03-16卢镇泽李凌崔玉真王伟杨清燕李黎娜蔡继福肖海兵
卢镇泽 李凌 崔玉真 王伟杨清燕李黎娜蔡继福肖海兵
克-雅病(Creutzfeldt-Jakob disease,CJD)临床上分为散发型、家族型、医源型和变异型。CJD早期临床表现为头痛、头晕、乏力、精神症状、认知功能减退,逐渐进展出现视觉障碍、肌阵挛、共济失调、锥体外系及锥体束损伤,晚期常处于昏迷或无动性缄默状态。CJD不同时期脑电图和核磁共振均有不同特征,通过长期随访动态观察脑电图及磁共振变化有助于临床医生更好认识CJD脑部结构及脑电的演变过程。我院2018年10月收治1例散发性CJD(sporadic Creutzfeldt-Jakob disease,sCJD)患者,截至写稿时已住院18个月。我们对该患者脑磁共振和脑电图进行了动态复查,结合临床资料,分析其不同时点的动态变化,特报告如下。
1 临床资料
患者女,71岁,因“头晕、右手抖动 1个月,右侧肢体无力20 d”于2018年10月入院。入院前1个月出现头痛、头晕,伴有右手不自主发作性痉挛抖动,20 d前出现右侧肢体乏力,持物不稳,行走拖步,外院颅脑MRI+DWI+MRA提示多发缺血病灶,予以缺血性脑血管病相关治疗无效。1周前出现行走、进食困难,言语含糊,呛咳,并出现双手发作性痉挛抖动,反应迟钝,答非所问,严重时言语不能、嗜睡。
体格检查:体温 36.8℃,脉搏:89次/min,血压145 mmHg/72 mmHg,GCS 9 分(E3V2M4),查体不合作,昏睡,意识模糊,混合性失语,时间、空间、人物定向力障碍,计算、执行不能完成,左侧肌张力增高,右侧肌张力正常,四肢肌力3~4级,双手发作性肌阵挛,双侧痛触觉对称,四肢腱反射活跃,双侧病理征阳性,颈软,脑膜刺激征阴性。
辅助检查:CSF常规、生化、细菌、真菌涂片及培养,CSF隐球菌抗原、单纯疱疹病毒、带状疱疹病毒、巨细胞病毒 DNA,结核 DNA未见异常。血及脑脊液寡克隆带、自身免疫性脑炎(NMDAR、AMPAR1/2、LGI1、CASPR2、GABA B Receptor、DPPX、IgLON5) 及副肿瘤抗体(Hu、Yo、Ri、CV2、PNMA2、amphiphysin、recoverin、SOX1、titin、Zic4、GAD65、Tr(DNER))阴性。血 HIV、梅毒血清学、乙肝两对半、ACTH、皮质醇、乳酸、维生素B12、血氨、糖化血红蛋白、甲状腺功能七项、ANA、ENA+dsDNA、ANCA、血清蛋白电泳、免疫固定电泳未见异常。
诊疗过程:结合患者症状、体征及相关辅助检查,初步诊断考虑脑炎可能(具体病因不明),入院第4天监测床边脑电图提示广泛尖波、尖慢波,第5天出现右侧面部及右上肢抽动,伴左上肢持续不自主抖动,随后意识障碍进展、呼之不应,诊断非惊厥癫痫持续状态,予以气管插管保护气道及抗癫痫药物治疗。排除急性感染,予以激素冲击(1 g/d,5 d)、免疫球蛋白[0.5 g/(kg·d),5 d]治疗脑炎,效果欠佳,仍处于浅昏迷状态。动态检测视频脑电图,发病2个月后监测到周期性三相尖慢复合波,患者此时处于持续浅昏迷状态,无肌阵挛发作(见图1)。
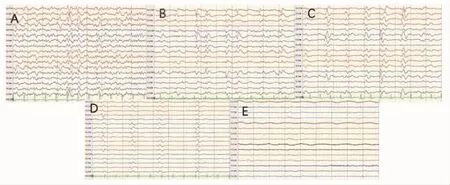
图1 脑电图 A:患者发病1个月的脑电图,表现为尖慢波;B:发病1个半月,表现为不典型周期波;C:发病2个月,表现为周期波(周期性渐趋严格,波形持续时间大约300 ms,波间间隔时间大约1000 ms);D:发病3个月,表现为周期波(与之前的脑电图形相比,波间间隔时间越来越长,大约1500 ms,周期性电位波幅较前下降);E:发病1年,表现为弥漫性低平慢波。
动态监测颅脑MRI,发病初期为双侧皮层、基底节区DWI高信号改变,病变进行性扩展,随后出现脑组织萎缩,发病12个月后复查可见广泛皮层萎缩、脑室扩大。T2可见双侧皮层及基底节区进行性萎缩、脑室扩大(见图2)。本例患者最终诊断:sCJD。
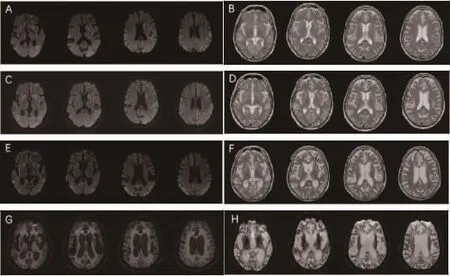
图2 sCJD患者得到磁共振演变 A~B:患者发病1个月的磁共振DWI及T2序列;C~D:分别为发病2个月DWI及T2序列;E~F:分别为发病8个月DWI及T2序列;G~H:分别为发病12个月DWI及T2序列,表现为明显的脑萎缩、侧脑室增大以及皮层脑脊液样增厚,称为皮层假性肥大(pseudohypertrophic cortex)[1]。
2 讨论
CJD是1920-1922年由Creutzfeldt和Jakob首次报告后命名,致病原因是人类朊蛋白异常变异,成为一种具有传播性的朊蛋白[3]。朊蛋白还可因其编码蛋白的PRNP基因突变引起遗传性或家族型朊蛋白病,以及Kuru病、Gerstmann-Straussler综合征、致死性家族性失眠症[4]。
sCJD(散发型克-雅病)是CJD最常见的类型,约占80%~90%,世界范围内发病率大概是百万分之一,在我国的好发年龄为55~75岁,男性发病率略高于女性,中位生存时间6~7个月,与欧洲相近。在另外一项日本研究中,近半数的sCJD患者生存时间超过1年,中位生存时间为12.9个月[5-7]。
sCJD早期-临床诊断困难,需要-结合颅脑影像学、脑电图、基因检测、脑脊液14-3-3蛋白或病理活检进一步确诊。受限于患者脑组织具有感染性,病理检查相当困难,14-3-3蛋白检查普及性也非常有限,因此颅脑磁共振及脑电图,对于诊断具有重要指导意义。目前多数文献报告展示的是疾病某一个时期磁共振或脑电图的特点。联合脑电图及磁共振的动态变化的报告少见,我们报告的病例时间跨度有18个月,磁共振及脑电图呈现疾病的动态进展。
sCJD存在较特异性的颅脑MRI影像学改变,在T2WI和DWI序列中可出现基底节核团区域、丘脑和广泛皮层高信号改变,无增强病灶,此类病变发病早期可为单侧,亦可累及双侧、广泛存在,这些特殊的影像改变被分别称为曲棍球征(累及丘脑枕与丘脑背内侧)和绸带征(累及皮层)[8-9]。我们团队既往研究发现,在诸如抗γ氨基丁酸B型受体(GABABR)脑炎等自身免疫性脑炎患者中也会出现皮层绸带征的现象[10-11],但我们仍然认为,这种绸带征与相应临床和脑电图表现合并出现时,对CJD有高度特征性的提示价值。部分患者在病程后期的磁共振呈现弥散性皮质萎缩,侧脑室扩大,形成皮层假性肥大(Pseudohypertrophic cortex)[1,12-13]。
EEG对诊断 sCJD具有重要意义的,但相对颅脑磁共振DWI序列和脑脊液14-3-3蛋白来说,敏感性稍低。在sCJD的病程中,大约三分之二的患者出现过周期性尖慢复合波(PSWC)[14-16],因此脑电图在 1998年被世界卫生组织列入sCJD的诊断标准中。在病程的任一时期均可出现典型PSWC,如高幅双相、三相或多相尖波、尖慢综合波,其中以三相波为主,持续时间为100~600 ms,每隔0.5~2 s重复一次[17]。在sCJD的早期或者晚期,脑电图记录以非特异性脑电图为特征,例如以弥漫性慢波为背景活动的表现,而在病程中动态监测脑电图,当出现PSWC时,具有诊断意义[18]。有研究证明,EEG上出现PSWC的特异性达到91%(最终为组织活检确诊sCJD),阳性预测值达到95%[10]。而在另外一项研究中,可能或很可能是CJD的805例患者中,PSWC特异性达到74%,阳性预测值达到93%[19]。
鉴于sCJD是一种不可治疗的疾病,诊断该病一定要谨慎。在疾病早期,临床表现、影像学、脑电图等结果不支持CJD诊断时,一定不要放弃其他可治性疾病的诊断,例如病毒性或自身免疫性脑炎、中毒或代谢性脑病等。在疾病演变到影像学和脑电图学均支持CJD诊断之时,且谨慎排除了可逆性疾病之后,可以坦诚与家属沟通,详细交代预后,进行营养支持对症治疗及人文关怀。
本例患者具有典型的EEG及MRI动态改变,脑电图动态演变的周期性尖慢复合波、早期磁共振皮层、基底节区DWI高信号改变以及晚期磁共振皮层假性肥大(pseudohypertrophic cortex)均具有较高特异性,从 2018年美国CDC的诊断标准[2]来说,患者有神经精神障碍,快速进展的认知障碍、肌阵挛、锥体束、锥体外系症状,脑电图有周期性尖慢复合波,颅脑磁共振成像多皮层及尾状核/壳核在DWI呈高信号,符合很可能的CJD诊断,但是从动态改变及特异的影像结果来说,即使缺乏脑脊液或病理组织的活检,确诊CJD的可能性非常大,这一结果提示我们,通过动态监测EEG及颅脑MRI等无创检查,也有可能确诊CJD。
本文报告1例sCJD患者,通过相关资料,我们认识到该病的脑电图呈现一个动态演变,随着疾病的发展可以出现典型的周期性尖慢复合波,具致病性的朊蛋白早期累及皮层及基底节区,随着疾病的进展,逐步扩散到相关区域和组织,形成磁共振具有特异性的皮层假性肥大。这类疑难罕见疾病的初期诊断往往困难,但通过动态复查脑电图及磁共振,如发病 1、2、3、6、12个月复查,若发现典型的动态影像学改变,即使在缺乏脑脊液14-3-3蛋白检测或病理组织的活检下,对于诊断CJD的意义也非常大。希望通过这个病例报告,可以让神经内科同道更深刻地了解该病进展过程中的辅助影像特点,从而提升该病的诊治水平。
