胡麻株高QTL定位与候选基因功能分析
2020-03-14宋夏夏王利民张建平张天豹刘彩月龙艳裴新梧
宋夏夏, 王利民, 张建平, 张天豹, 刘彩月, 龙艳*, 裴新梧
(1.中国农业科学院生物技术研究所, 北京 100081; 2.甘肃省农业科学院作物研究所, 兰州 730070)
亚麻(LinumusitatissimumL.)属于亚麻科亚麻属,为一年生草本植物。亚麻一般可以分为纤维亚麻、油用亚麻和兼用亚麻。胡麻为油用亚麻,是我国华北和西北地区重要的油料作物和经济作物。随着人们生活水平的提高,绿色健康的食用油越来越受欢迎。胡麻是α-亚麻酸含量较高的油料作物之一,其籽油中α-亚麻酸的含量约占总脂肪酸的50%,使得胡麻籽油在食品、饲料和工业应用中具有重要价值[1-2]。α-亚麻酸是人体内长不饱和脂肪酸EPA(eicosapentaenoic acid,二十碳五烯酸)、DPA(docosapentaenoic acid,二十二碳五烯酸)和DHA(docosahexaenoic acid,二十二碳六烯酸)的前体,具有降血脂、预防心血管疾病、抗炎抗癌和改善记忆力等保健功效[3-5]。同时,胡麻籽还含有胡麻胶、木酚素、膳食纤维等对人体有益的物质[6],在食品保健方面具有广阔的开发前景。株高不仅影响胡麻产量和胡麻籽品质,还与植株倒伏性状相关,是一个由基因和环境共同作用的复杂数量性状(quantitative trait loci,QTL)。因此,研究控制胡麻株高的QTL有助于了解胡麻生长的分子基础和调控机理,为胡麻品种的性状改良和产量提高奠定基础。快速发展的高通量测序技术和有效的植物基因组参考序列为高分辨率性状作图和快速识别候选基因或诊断标记提供了可能性。
QTL-seq技术充分利用BSA和高通量测序技术,直接将具有差异性状的基因池混合进行高通量测序,获得大量丰富的SNP标记,计算每个SNP位点的SNP-index(该位点与参考序列不同的reads数占总reads数的比例),然后将两个基因池每个位点的SNP-index相减得到差值ΔSNP-index,从而获取全基因组范围内的ΔSNP-index图谱。由于除目标基因区域外的其他基因组区域理论上没有差异,即ΔSNP-index接近于0,所以将ΔSNP-index显著偏离0的位点作为候选位点。QTL-seq策略最先在水稻中提出,利用水稻F2和RIL群体成功定位了水稻中抗病及与幼苗活力相关的QTL[7]。Daware等[8]用QTL-seq结合SSR方法定位了调控水稻粒重的基因,构建了水稻高密度SSR图谱,包含3 791个标记,总图距为2 060 cM,标记间平均距离为0.54 cM。通过生物信息学分析,将控制粒重的QTL粗定位在6条染色体上,共解释31%的表型变异。而后结合QTL-seq和差异基因表达分析,缩小了一个粒重主效QTL的遗传区间,成功定位了一个粒重主效基因。Shu等[9]研究了花椰菜和甘蓝中控制开花时间的QTL。同时,该方法已成功应用于黄瓜早花QTL[10]、西红柿重量和果实数QTL[11]、鹰嘴豆百粒重和根源特质比相关QTL[12-14]以及木豆抗病性相关QTL[15]。
本研究以加拿大高含油量油用品种Macbeth和中国纤用品种黑亚14号为亲本构建的重组自交系(RIL F7)群体为材料,统计了该群体在不同时间不同地区的株高表型,利用QTL-seq方法对胡麻株高QTL进行了定位分析和候选基因预测,并将候选株高基因转入拟南芥中进行基因功能验证。本研究不仅初步鉴定了胡麻中控制株高的功能基因,同时探索出利用QTL-seq方法快速有效定位并克隆胡麻数量性状主效基因的方法。
1 材料与方法
1.1 材料
供试材料为Macbeth(母本)、黑亚14号(父本)及得到的RIL F7群体,共155个株系,由甘肃省农业科学院作物研究所提供。试验材料于2016年种植于中国农业科学院河北廊坊试验基地,每行种植一个株系30株,行宽1 m,行距20 cm。亲本及RIL群体各株系分别设置3个重复种植。种植期间进行常规田间管理,待至成熟期时统计单株株高。
1.2 方法
1.2.1株高统计和分析 在胡麻成熟期,测量和统计地面到植株顶端高度(cm)。每一个RIL株系以3株植株的株高均值作为表型值。结合该群体在甘肃兰州、甘肃景泰和云南元谋3个不同环境的株高数据(甘肃省农业科学院作物研究所提供),比较不同环境的各单株的株高表型。选取在四个环境均表现为高、低株的株系各15株分别作为高、低株混池备选单株。
1.2.2胡麻基因组DNA的提取 在苗期取2个亲本及RIL群体所有单株的叶片,采用植物基因组DNA提取试剂盒(北京全式金生物技术有限公司)提取基因组DNA,用琼脂糖凝胶电泳进行完整性检测。
1.2.3文库构建及测序 检测合格的DNA加灭菌ddH2O稀释,将浓度统一调整为100 ng·μL-1。将2个池的各15个株系单株DNA等体积混合,获得高株基因池和低株基因池。采用TruSeq Library Construction Kit构建亲本和两个基因池文库,通过Illumina HiSeqTM PE 150进行测序。文库构建完成后,先使用Qubit 2.0进行初步定量,稀释至1 ng·μL-1,随后利用Agilent 2100对文库的插入大小进行检测,符合预期后使用QPCR方法[16]对文库的有效浓度(>2 nmol·L-1)进行准确定量,以保证文库质量。库检合格后进行Illumina HiSeq TM PE150测序。
1.2.4生物信息分析 对测序得到的原始数据(raw data)进行质控得到可用数据(clean data);根据比对结果,进行SNP、InDel的检测及注释,从而得到每个样本的SNP数据,初步确定株高候选基因。
1.2.5株高候选基因植物表达载体构建 根据植物表达载体35s-redkan的多克隆位点AseⅠ和BamHⅠ两侧同源序列,设计带有载体同源序列的引物(LuCWINV1-Fi:5’-ACGCGTAAGGGG-ATCCGGTGCACAATGGAATTTACCA;LuCWINV1-Ri:5’-CGGGTCTAGAGAATTCGGTTCACCATTCG-GGAGTATC)用于扩增包含载体接头的LuCWINV-1序列。采用Minibest Plant RNA Extraction试剂盒(TaKaRa)提取亲本总RNA,反转录得到cDNA后进行扩增。扩增体系:LuCWINV1-Fi/Ri引物各0.5 μL(10 μmol·L-1),模板2 μL,2×EasyTaqPCR SuperMix 25 μL,ddH2O 22 μL。反应程序:94 ℃预变性3 min;94 ℃变性10 s,56 ℃退火5 s,72 ℃延伸5 min,35个循环;72 ℃再延伸5 min,4 ℃保存。经10 g·L-1琼脂糖凝胶电泳检测扩增产物。PCR产物和酶切产物分别经切胶纯化后,以35s-redkan植物表达载体,用In-Fusion HD Cloning Kit连接并转化大肠杆菌,用含有50 mg·L-1Kan的LB平板筛选阳性克隆。
1.2.6拟南芥的遗传转化 利用冻融法将构建好的载体转入农杆菌EH105中,采用蘸花法进行拟南芥的遗传转化[17]。构建好的植物表达载体35S-Redkan-LuCWINV1-1Hei和35S-Redkan-LuCWINV1-1Mac具有红光选择标记,在绿色光源下,用红色眼镜观察收获的转基因种子会发出红光。在拟南芥培养室中种植转基因拟南芥阳性株系,观察T3代纯合株系的表型。当拟南芥生长至株高不再增加时,测量其株高。
2 结果与分析
2.1 重组自交系系株高统计和分析
结合甘肃兰州和景泰及云南元谋的三个地区株高数据,分析不同环境下的RIL群体株高分布情况。结果表明,在4个环境中,黑亚14号株高均比Macbeth高,且在4个环境中的RIL群体株高表型分布均为正态分布(图1)。选取4个环境中均表现为高株和低株的胡麻株系各15株分别作为高/低单株池,用于胡麻株高QTL定位。
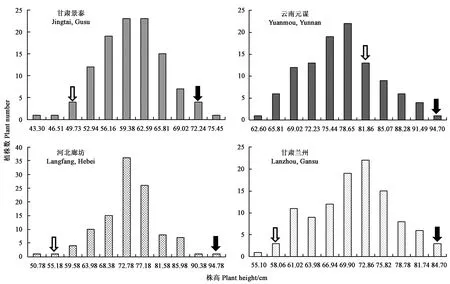
注:实心和空心箭头分别为父本黑亚14号和母本Macbeth。
2.2 测序结果分析和初步确定株高候选基因
对胡麻RIL群体高、低株混池及父、母本进行重测序,检测变异位点。结果显示,测序样品在胡麻基因组上覆盖率为84.62%~89.71%,测序平均覆盖深度为13.15~15.39(表1)。据统计,基因上发生SNP位点突变的总数量为362 347个,其中SNP发生在基因上游的有29 492个,发生在外显子上的有45 418个,发生在内含子上的有44 616个,发生在可变剪接区的有138个,发生在基因下游的有27 319个,发生在基因间隔区的有209 051个。发生在外显子上的突变包括导致翻译提前终止的有348个,导致翻译延后终止的有98个,同义突变22 602个以及非同义突变22 370个(表2)。
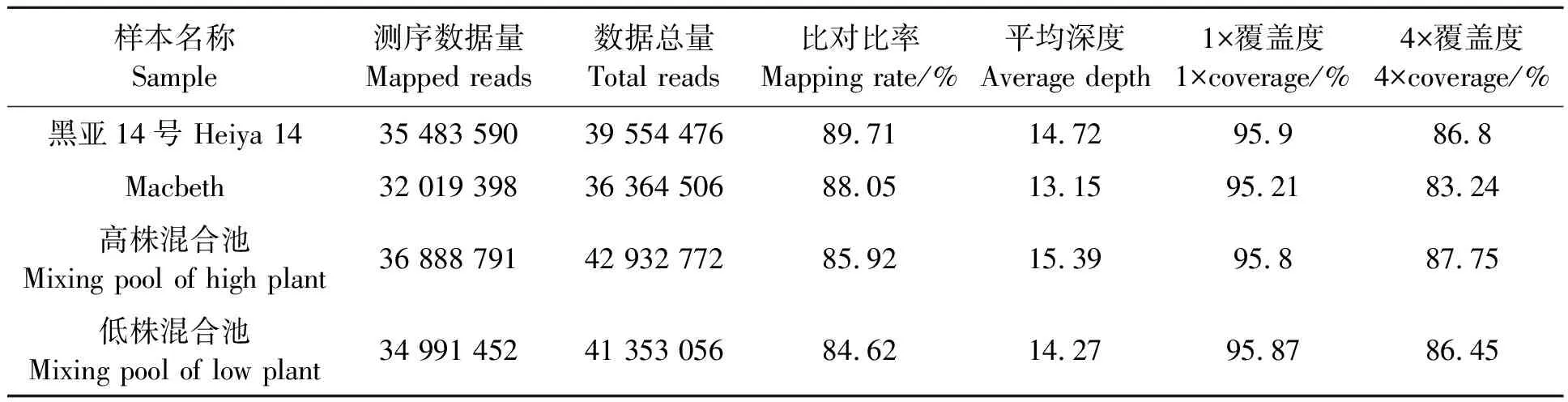
表1 测序深度及覆盖度

表2 SNP检测及注释结果
本研究将QTL-seq方法得到的结果与前期传统QTL定位结果[18]结合起来分析发现,位于14连锁群上的主效QTL定位在scaffold 603上,同时在delta-SNP index上分析发现位于scaffold 603上的基因Lus10015614在其上游区(49 861 bp)存在SNP位点,将该基因作为候选基因。
2.3 株高主效基因的克隆分析
从phytozome数据库中下载得到Lus10015614的基因组和cDNA序列。该基因序列与拟南芥At3g13790(Atwinv1)匹配度非常高(67%),拟南芥At3g13790为细胞壁转化酶(cell wall invertase),将胡麻中lus10015614.g命名为LuCWINV1-1。phytozome数据库中LuCWINV1-1的基因长度为3 818 bp,编码区长度为1 749 bp。设计引物在黑亚14(父本)和Macbeth(母本)的LuCWINV1的基因组和cDNA中扩增,得到了和目标大小相同的片段,测序后对序列进行分析。
测序结果显示,黑亚14号和Macbeth中该基因的长度均为3 818 bp,但是序列不完全一致,两个亲本的氨基酸序列共有2个氨基酸发生改变,导致Macbeth中第21位的谷氨酸和第523位异亮氨酸在黑亚14中分别为甘氨酸和丙氨酸。
2.4 黑亚14号和Macbeth中LuCWINV1-1不同时期内源表达分析
为了研究LuCWINV1-1在胡麻中的表达模式,本研究选取不同时期和不同组织的RIL群体亲本胡麻植株进行荧光定量分析。胡麻在第3~7周为快速生长期,第7周处于快速生长期后期。结果显示,生长2~6周的黑亚14号和Macbeth的叶片和茎中LuCWINV1-1的表达量处于较低水平,同时在这两个品种之间没有很大差异。在胡麻快速生长后期(第7周)和成熟期(第8和9周),LuCWINV1-1的表达量大幅增加。此外,黑亚14号在第9周时与之前相比叶中LuCWINV1-1的表达量显著增加,但是不如Macbeth的叶片中同一时期的表达量高(图2)。
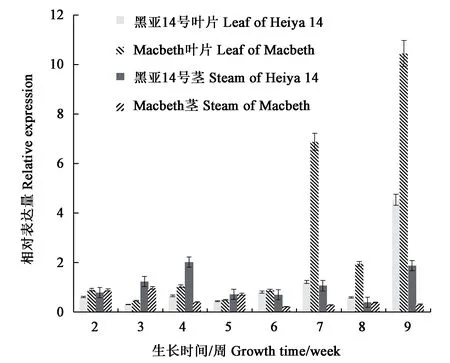
图2 黑亚14号和Macbeth不同组织不同时期LuCWINV1-1表达量
2.5 株高候选基因在拟南芥中的遗传转化和功能验证
将胡麻黑亚14号和Macbeth中的LuCWINV1-1基因通过农杆菌介导的蘸花法遗传转化拟南芥,获得T3代纯合转基因株系,以野生型拟南芥为对照比较株高表型。
统计结果表明,与野生型拟南芥相比,4个LuCWINV1-1Mac转基因株系(6F-1、6F-2、6F-4和6F-6)的株高表现为极显著降低;4个LuCWINV1-1Hei转基因株系(6M-10、6M-11、6M-12和6M-14)的株高表现为极显著增加,1个LuCWINV1-1Hei转基因株系(6M-13)表现为显著增加(图3,表3)。综上所述,LuCWINV1-1对胡麻株高的解析具有重要意义,可用于后续研究。
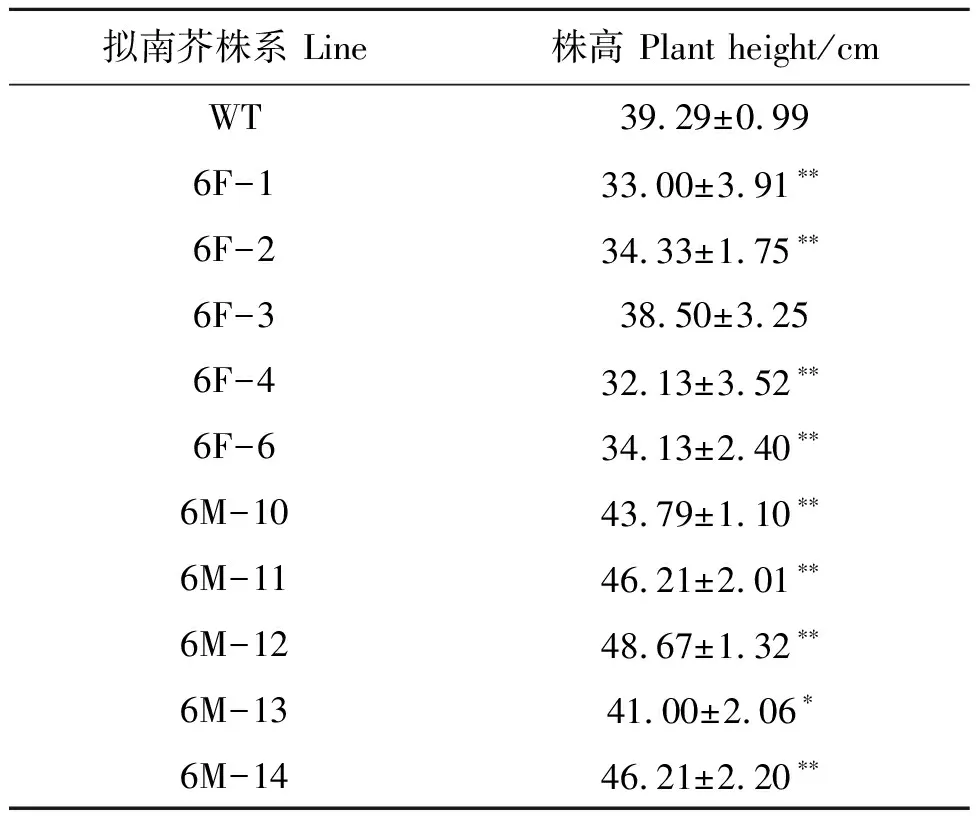
表3 转基因拟南芥和野生型株高统计

图3 LuCWINV1-1Hei和LuCWINV1-1Mac转基因株系表型
3 讨论
QTL-seq是植物中快速有效定位QTL的方法之一,随着高通量测序技术的快速发展和测序价格的降低,近年来已成功应用于多种植物中。胡麻为二倍体作物,基因组相对较小,但目前还没有QTL-seq方法应用于胡麻QTL研究的相关报道。本研究以胡麻RIL群体(Macbeth×黑亚14号)为材料,利用QTL-seq成功检测了控制胡麻株高性状的QTL。在scaffold2096、scaffold2057和scaffold228等10个不同的scaffold上共检测到10个与株高有关的QTL。本研究根据群体内各株系的株高表型数据构建两个极端表型的混池,将QTL-seq方法应用于胡麻QTL的定位研究中。由于作物数量性状易受环境影响,本研究结合多年多点的表型数据进行基因池的构建,排除了环境的干扰,提高了QTL定位的精度。在利用测序QTL-seq寻找差异SNP位点时,测序数据的深度及基因组覆盖度为能否找到合适SNP位点的关键因素。平均测序深度达到10×以上,能有效找到合适的SNP位点。因此,在不同作物利用QTL-seq方法进行SNP关联位点分析时要根据基因组倍性、大小等因素来设计合适的测序倍数来进行。本研究结果表明,QTL-seq应用于胡麻QTL定位研究的可行性,为以后快速定位控制胡麻重要性状的基因提供了基础。
通过与拟南芥、水稻基因组序列进行比较发现,本研究定位的LuCWINV1-1属于酸性转化酶中的细胞壁转化酶,包含细胞壁转化酶基因保守的结构域NDPNG、RDP和MWECP。胡麻RIL群体亲本黑亚14号和Macbeth中的LuCWINV1-1氨基酸序列不同,这预示着等位基因功能的变化可能是由于氨基酸的改变造成的。拟南芥转基因植株株高表型也初步证明了这一推断,来自于Macbeth的等位基因导入后植株株高明显低于由黑亚14号的等位基因导入后植株的株高。在拟南芥中也发现,转化酶家族与植株生长发育有关,转化酶的缺失会导致拟南芥生长发育速度减慢,根长变短,植株变矮,发芽和开花受到影响,植株生物量减少[19-23],本研究所鉴定的胡麻LuCWINV1-1基因具有潜在研究价值。
