以amdS为筛选标记的黑曲霉遗传转化体系的建立
2020-03-13孙菁任文琪许勤虎曹威刘浩
孙菁, 任文琪, 许勤虎, 曹威*, 刘浩*
(1.天津科技大学生物工程学院, 工业发酵微生物教育部重点实验室, 天津市微生物代谢与发酵过程控制技术工程中心, 天津 300457; 2.天津实发中科百奥工业生物技术有限公司, 天津 300462)
黑曲霉是一种重要的工业发酵微生物,属于公认安全(generally regarded as safe, GRAS)的食品级微生物,可利用廉价的可再生碳源,与其他细胞工厂相比具有显著的优势,被广泛用于发酵生产多种有机酸和酶制剂[1-3]。通过遗传代谢工程手段对黑曲霉菌株开展研究,不仅可以更好地理解其代谢调控机制,还可以有目的地进行遗传改造,提高目标产物的产量,优化菌株的发酵性能。遗传改造过程中,筛选标记基因必不可少。当前常被用于黑曲霉遗传操作的筛选标记基因非常有限,如博来霉素抗性基因ble[4]、潮霉素B抗性基因hph[5]、乳清苷酸脱羧酶编码基因pyrG[6]、鸟氨酸氨甲酰基转移酶基因argB[7]等。复杂的代谢工程改造往往涉及诸多基因的敲除或表达,有限的筛选标记基因难以满足需要。
amdS是构巢曲霉中乙酰胺酶编码基因,赋予其可在乙酰胺作为唯一氮源的培养基中生长的能力,其表达调控机制已被广泛研究[8-9]。Hynes等[9]发现,黑曲霉因缺乏amdS基因而不能利用乙酰胺作为唯一氮源,因此,amdS可作为黑曲霉转化过程中的筛选标记。
在黑曲霉发酵生产柠檬酸过程中,草酸是一种主要的副产物。微生物中草酸的合成途径主要有两种:一是乙醛酸代谢中,乙醛酸在乙醛酸氧化酶(EC 1.2.3.5)作用下生成草酸;二是草酰乙酸在草酰乙酸乙酰基水解酶(OAH,EC 3.7.1.1)作用下水解生成草酸和乙酸,而在黑曲霉中只存在草酰乙酸乙酰基水解酶编码基因oahA。在α-淀粉酶生产菌株黑曲霉BO-1中,敲除OAH编码基因后发酵液中不再含有副产物草酸[10]。因此,黑曲霉中草酸的生物合成只来源于OAH对草酰乙酸的水解过程[10-11]。本研究在黑曲霉中成功构建了以amdS基因为营养筛选标记的遗传转化系统,赋予其对乙酰胺的利用能力,为转化子的筛选提供了正向选择标记,并应用该筛选标记成功获得黑曲霉oahA敲除菌株,同时对其合成草酸水平的变化进行了分析。
1 材料与方法
1.1 试验材料
1.1.1菌株和质粒 大肠杆菌JM109由天津科技大学工业发酵微生物教育部重点实验室保藏。黑曲霉ATCC1015(编号S304)购自广东省微生物菌种保藏中心。构巢曲霉(编号S478)由中国科学院微生物研究所提供;质粒pLH331为表达质粒(含有loxP-hph-loxP元件及两侧的多克隆位点)用于oahA基因回补菌株构建;质粒pLH334为构建敲除质粒出发载体(含有ble标记基因,在loxP-hph-loxP元件两端含有多克隆位点)由本实验室构建保存。宿主菌中含有可被多西四环素诱导的Tet-On系统,能够介导重组酶Cre的表达。潮霉素B抗性筛选标记hph两侧包含两个同向的loxP位点,在Cre重组酶的作用下hph从基因组上被切除。
1.1.2主要仪器与试剂 RH-250-A型生化培养箱购自韶关市泰宏医疗器械有限公司;HNY-200B型控温摇床购自上海志成设备厂;1260系列高效液相色谱仪(HPLC)购自安捷伦科技有限公司;SCD-II型高纯水装置购自军事医学科学院卫生装备研究所;TGL-16台式高速离心机购自上海医用分析仪器厂;EDC-810型PCR仪购自德国Eppendorf公司;4N75光学显微镜购自尼康公司。
限制性内切酶购自TaKaRa公司,总RNA提取试剂盒和反转录试剂盒购自天根生化科技(北京)有限公司,Phanta Max高保真酶购自北京全式金生物技术有限公司,PrimeSTAR HS(Premix)高保真酶购自宝日医生物技术(北京)有限公司,ClonExpress II One Step Cloning Kit购自南京诺唯赞生物科技有限公司,PCR引物(表1)由华大基因股份有限公司合成。卡那霉素购自生工生物工程(上海)股份有限公司,乙酰胺购自上海阿拉丁生化科技股份有限公司,其他试剂均为国产分析纯。

表1 本研究中所用引物
1.1.3培养基 LB培养基:NaCl 10 g·L-1,胰蛋白胨10 g·L-1,酵母提取物5 g·L-1,121 ℃灭菌20 min。固体培养基中加入琼脂粉15 g·L-1。
PDA培养基:去皮马铃薯200 g,切成小块,加适量蒸馏水煮沸约30 min,纱布过滤,取清液。加入20 g葡萄糖完全溶解,然后加蒸馏水定容至1 L,121 ℃灭菌20 min。固体培养基加入琼脂粉15 g·L-1。
真菌生长完全培养基(CM):取蒸馏水897 mL加入琼脂15 g,121 ℃灭菌20 min。依次加入ASP+N 20 mL,CM Trace elements 1 mL,10%酪蛋白水解物10 mL,50%葡萄糖20 mL,10%酵母浸出物50 mL,1 moL·L-1MgSO42 mL。其中,ASP+N:氯化钾2.61 g,磷酸二氢钾7.48 g,硝酸钠29.75 g,pH 5.5,加蒸馏水定容至100 mL,121 ℃灭菌20 min;CM Trace elements:ZnSO4·7H2O 2.1 g,H3BO31.1 g,MnCl2·4H2O 0.5 g,FeSO4·7H2O 0.5 g,CoCl2·6H2O 0.17 g,CuSO4·5H2O 0.16 g,Na2MoO4·2H2O 0.15 g,EDTA 5.1 g,ddH2O定容至100 mL,121 ℃灭菌20 min。
诱导培养基(IM):蒸馏水900.7 mL,121 ℃灭菌20 min。加入50%甘油10 mL,K buffer 0.8 mL,0.01% FeSO410 mL,MN buffer 20 mL,1% CaCl2·2H2O 1 mL,1 moL·L-1MES 40 mL,IM Trace elements 5 mL,20% NH4NO32.5 mL,20%葡萄糖10 mL。其中,K buffer:KH2PO4(1.25 mol·L-1)17.01 g加蒸馏水定容至100 mL;K2HPO4(1.25 mol·L-1)21.77 g加蒸馏水定容至100 mL;将K2HPO4加入到KH2PO4中并调节pH至4.8,121 ℃灭菌20 min。MN buffer:MgSO4·7H2O 3 g,NaCl21.5 g,蒸馏水定容至100 mL,121 ℃灭菌20 min。IM Trace elements:ZnSO4·7H2O 10 mg,CuSO4·5H2O 10 mg,H3BO310 mg,MnSO4·H2O 10 mg,Na2MoO4·2H2O 10 mg,加蒸馏水定容至100 mL,121 ℃灭菌20 min。固体培养基中20%葡萄糖5 mL,添加琼脂粉15 g·L-1。
黑曲霉液体培养基:MgSO4·7H2O 1 g·L-1,葡萄糖20 g·L-1,KH2PO41 g·L-1,酵母浸出物10 g·L-1,调节pH至4.0,115 ℃灭菌20 min。
黑曲霉草酸发酵培养基:葡萄糖30 g·L-1,K2HPO41 g·L-1,MgSO4·7H2O 0.5 g·L-1,ZnCl20.001 g·L-1,MnSO4·5H2O 0.014 g·L-1,(NH4)2SO43 g·L-1,KH2PO41 g·L-1,FeCl3·6H2O 0.01 g·L-1,MES 0.5 mol·L-1,调节pH至4.0,115 ℃灭菌30 min。
基本培养基(MM):无机盐溶液 20 mL,D-葡萄糖10 g·L-1,乙酰胺10 mmol·L-1,定容至1 L。其中无机盐溶液:KCl 26 g,MgSO4·7H2O 26 g,KH2PO476 g,微量元素溶液50 mL,加蒸馏水定容至1 L;微量元素溶液:Na2B4O7·10H2O 40 mg,CuSO4·5H2O 400 mg,FePO4·2H2O 800 mg,MnSO4·2H2O 800 mg,Na2MoO4·2H2O 800 mg,ZnSO4·7H2O 8 g,加蒸馏水定容至1 L。
1.2 试验方法
1.2.1黑曲霉基因组DNA的提取 收集相应菌株的菌丝体,用蒸馏水洗涤2~3次后经液氮研磨至粉状,取约200 mg粉末于2 mL EP管中,加入950 μL的DNA提取缓冲液(NaCl 100 mmol·L-1,EDTA 50 mmol·L-1,SDS 1%,Tris 50 mmol·L-1,pH 8.5)重悬并于65 ℃下放置30 min。加入350 μL 5 mol·L-1的醋酸钾振荡混匀,于-20 ℃下静置15 min,4 ℃ 12 000 r·min-1离心,取1 mL上清液并加入等体积苯酚混合物,剧烈混匀,4 ℃ 12 000 r·min-1离心,收集水相并逐滴加入350 μL异丙醇,4 ℃ 12 000 r·min-1离心收集沉淀基因组DNA。
1.2.2基因的PCR扩增 根据NCBI中构巢曲霉amdS表达盒序列,分别设计上下游引物P1055/P1056,以构巢曲霉基因组DNA为模板扩增amdS表达盒序列。PCR体系:模板DNA 1 μL,dNTP Mix(10 mmol·L-1)1 μL,2×Phanta Max buffer 25 μL,引物各 2 μL,Phanta Max 1 μL,ddH2O 18 μL,总体积50 μL。反应条件:95 ℃预变性1 min;95 ℃变性15 s,60 ℃退火15 s,72 ℃延伸3 min 30 s,35个循环;72 ℃延伸5 min,得到3 368 bp的DNA序列。
以黑曲霉ATCC1015基因组DNA为模板,以P953/P954为引物分别扩增oahA的上游片段,以P955/P956为引物扩增oahA的下游片段。PCR体系:模板DNA 2 μL,引物各 2 μL,Premix 25 μL,ddH2O 19 μL,总体积50 μL。反应条件:98 ℃预变性3 min;98 ℃变性10 s,60 ℃退火5 s,72 ℃延伸1 min 15 s,35个循环;72 ℃延伸10 min,分别得到1 025 和1 049 bp的DNA序列。
以黑曲霉ATCC1015基因组DNA为模板,以P816/P817为引物扩增含有其自身启动子的oahA基因表达盒序列。PCR体系:模板DNA 1 μL,dNTP Mix(10 mmol·L-1)1 μL,2×Phanta Max buffer 25 μL,引物各 2 μL,Phanta Max 1 μL,ddH2O 18 μL,总体积50 μL。反应条件:95 ℃预变性1 min;95 ℃变性15 s,60 ℃退火15 s,72 ℃延伸2 min 30 s,35个循环;72 ℃延伸5 min,得到2 457 bp的DNA序列。
1.2.3敲除质粒和回补质粒的构建 以本课题组pLH334作为出发载体,利用KpnⅠ/EcoRⅠ分别双酶切pLH334质粒和amdS表达盒序列片段,将pLH334上的ble筛选标记替换为amdS,得到新的基因敲除出发质粒pLH439。
将扩增的oahA上游片段(1 025 bp)经限制性内切酶酶切回收后,连接到pLH439质粒得到含有oahA上游片段的质粒pLH440;再将扩增得到的oahA下游片段(1 049 bp)经限制性内切酶酶切回收后,连接到pLH440质粒的SpeⅠ/HindⅢ酶切位点,得到包含oahA基因上下游片段的基因敲除质粒pLH453。
以pLH331作为出发载体,利用EcoRⅠ/BamHⅠ分别酶切pLH331质粒和oahA表达盒序列片段(2 457 bp),经T4连接酶连接重组后获得oahA基因表达回补质粒pLH372。
1.2.4农杆菌的电转化 将相应重组质粒与农杆菌感受态细胞混合后放入2 mm电击杯中,设置电压2.5 kV,电容25 μF,电阻率200 Ω,脉冲5 ms进行电击转化。然后迅速加入预冷的1 mL LB培养基,28 ℃,150 r·min-1培养6 h,取100 μL涂于含100 μg·mL-1卡那霉素的LB固体平板上,2 d后随机挑选圆形米白色单菌落进行PCR验证。
1.2.5农杆菌介导黑曲霉转化及转化子的筛选
将农杆菌单菌落接种于3 mL含有100 μg·mL-1卡那霉素的LB液体培养基中,28 ℃ 200 r·min-1振荡培养20 h。培养液经4 000 r·min-1离心10 min,菌体沉淀转接至5 mL的IM液体培养基中诱导培养6 h,取农杆菌与黑曲霉孢子1∶1混合液400 μL缓慢滴加到铺有0.45 μm滤膜的IM平板上,待风干后,将平板放置于25 ℃培养箱中培养2~3 d。加入0.6 mL无菌水将滤膜上的菌体洗涤下来,转移至含有100 μg·mL-1氨苄霉素、100 μg·mL-1链霉素、200 μg·mL-1头孢噻肟钠、250 μg·mL-1潮霉素B的CM平板上,28 ℃培养3~5 d,挑取单菌落接至含有250 μg·mL-1潮霉素B的PDA平板和含有10 mmol·L-1乙酰胺的MM平板中。根据需要表型挑取单菌落进行PCR验证。
1.2.6抗性筛选标记hph的切除 为切除转化子中的潮霉素B抗性筛选标记基因hph,收集相应菌株的孢子(约200个)涂布于含有30 μg·mL-1多西四环素的MM固体平板上培养5 d。多西四环素诱导Cre表达,位于两个loxP位点之间的hph将被重组切除。将长出的重组子分别点接在PDA培养基和含有250 μg·mL-1潮霉素B的PDA上,选取在PDA培养基上生长,而在含有潮霉素B的PDA培养基上不生长的克隆,提取其基因组DNA,以引物对P607/P608扩增hph基因。
1.2.7黑曲霉生长表型分析 为了考察黑曲霉转化子对乙酰胺的利用情况,分别将黑曲霉野生型菌株S304、含有amdS表达盒的黑曲霉转化子S665以及构巢曲霉菌株S478点接于含不同氮源的MM平板上。其中A培养基是含1%葡萄糖,0.2%硝酸铵和12 mmol·L-1CsCl的MM固体培养基,B培养基是含1%葡萄糖,10 mmol·L-1乙酰胺和12 mmol·L-1CsCl的MM固体培养基。28 ℃培养3~5 d后观察菌体生长情况。
为考察黑曲霉ΔoahA菌株及回补菌株的生长情况,分别点接黑曲霉野生型菌株S304、ΔoahA菌株S668和oahAΔoahA菌株S430于PDA培养基上,28 ℃培养3~5 d后观察菌体生长及产孢情况。
1.2.8黑曲霉中oahA基因表达水平分析 点接相应菌株于PDA平板上,28 ℃培养4~5 d长出足量新鲜孢子后向平板中加入3 mL无菌水,用棉签轻轻摩擦菌体表面,收集孢子。孢子悬液经Miracloth滤布过滤后用血球计数板进行计数。以2×106个·mL-1的孢子浓度接种于50 mL黑曲霉草酸发酵培养基中,28 ℃、200 r·min-1震荡培养3 d,12 000 r·min-1离心2 min,收集菌丝体并用无菌滤纸吸干,液氮研磨后提取总RNA,按照反转录试剂盒说明进行反转录PCR获得cDNA。设计引物P1002/P1003,分析黑曲霉ΔoahA菌株和oahA回补菌株中oahA基因转录水平的变化。以P992/P993为引物,肌动蛋白基因act1作为内参基因。PCR反应体系:模板cDNA 1 μL,相应引物各0.4 μL,Taq1.6 μL,ddH2O 14.5 μL,总体积20 μL。反应条件为:94 ℃预变性10 min;94 ℃变性30 s,55 ℃退火30 s,72 ℃延伸23 s,30个循环;72 ℃后延伸10 min。
1.2.9黑曲霉中草酰乙酸水解酶活性分析 收集在黑曲霉草酸发酵培养基中生长3 d的菌丝体,利用10 mmol·L-1的pH 7.0的磷酸钾缓冲液清洗三次。取约0.5 g菌体经液氮冷冻后研磨,粉末中加1 mL细胞蛋白提取缓冲液(0.1 mmol·L-1的MnCl2,50 mmol·L-1β-巯基乙醇,7%(质量体积分数)蔗糖,20 mmol·L-1Tris-HCl,pH 8.0)于0 ℃悬浮,12 000 r·min-1离心5 min后取上清液,用于总蛋白和酶活检测。胞内总蛋白的测定遵循标准考马斯亮蓝法,草酰乙酸水解酶酶活测定以草酰乙酸为底物,检测酶解后乙酸的生成量[13]。
1.2.10菌体干重和草酸产量测定 以2×106个·mL-1孢子浓度接种于50 mL黑曲霉草酸发酵培养基中,28 ℃、200 r·min-1培养3 d。期间每天取样,4 ℃、12 000 r·min-1离心2 min,收集上清液并进行适度稀释。将1 mL稀释液经0.22 μm滤器过滤后,使用安捷伦1260系列高效液相仪检测发酵液中草酸浓度。色谱检测条件:HPX-87H(Bio-Rad)有机酸柱,流动相:5 mmol·min-1H2SO4,流速:0.6 mL·min-1,柱温度65 ℃,UV 210 nm,检测时间15 min。同时,收集的菌丝体经蒸馏水洗涤2次后,于105 ℃干燥至恒重,计算菌体干重。
2 结果与分析
2.1 黑曲霉在含乙酰胺培养基上的生长情况
为了考察黑曲霉在含乙酰胺培养基上的生长情况,将野生型黑曲霉S304接入两种不同的培养基(图1)后发现,在含0.2%硝酸铵的MM培养基A中长出少量孢子,而在含10 mmol·L-1乙酰胺和12 mmol·L-1CsCl的MM培养基B中菌体不生长,无明显菌丝和孢子产生。而含amdS表达盒的黑曲霉转化子S665在B培养基中生长正常,且生长明显比出发菌株S304迅速。同时,构巢曲霉S478可以在含有乙酰胺的MM培养基上正常生长,这与先前报道一致[8-9]。因此,可以利用amdS基因作为黑曲霉转化筛选标记。
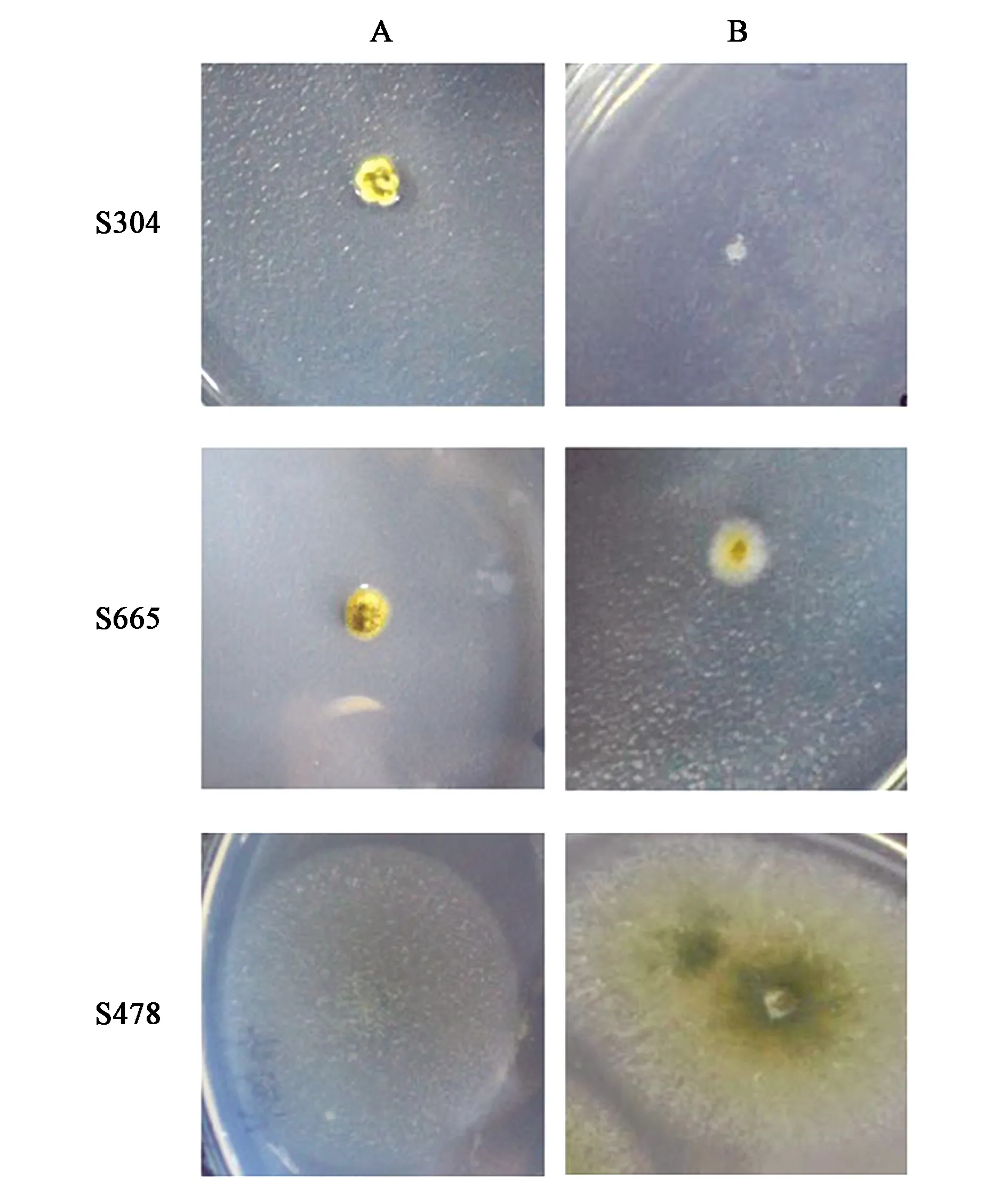
A:含1%葡萄糖,0.2%硝酸铵和12 mmol·L-1 CsCl;B:含1%葡萄糖,10 mmol·L-1乙酰胺和12 mmol·L-1 CsCl。
2.2 黑曲霉oahA敲除菌株中amdS筛选标记分析
将敲除载体pLH453通过电转化导入农杆菌感受态细胞中,挑取阳性克隆,与黑曲霉孢子进行共培养。从图2可以看出,amdS表达盒位于oahA上下游同源片段的外侧,当发生同源重组时,amdS被重组丢失而失去利用乙酰胺的能力,而位于oahA上下游片段之间的loxP-hph-loxP赋予菌体在含有潮霉素B培养基上的生长能力。将长出的转化子编号,依次转接至含有250 μg·mL-1潮霉素B的PDA抗性平板和含有10 mmol·L-1乙酰胺的MM筛选平板上。在挑取的41个转化子中,17号和21号转化子在含有潮霉素B的PDA平板上可以生长,而不能在含有乙酰胺的MM平板上生长(图2A)。因此,可初步判定这两株菌株为oahA敲除菌株。将17号和21号转化子提取基因组进行PCR验证分析。根据双交换的原理设计验证敲除的引物(图2C),若oahA被成功敲除,以转化子基因组DNA为模板,以P973/P974及P975/P976为引物扩增不出条带,而由于潮霉素B编码基因的存在,以P973/P641及P642/P976为引物则可以扩增出条带,同时以P973/P976为引物,扩增得到的片段比野生型菌株基因组为模板得到的片段长300 bp。其中,转化子17号与oahA敲除菌株预期情况一致,oahA敲除成功,命名为S668(图2C)。由此可知,利用此转化系统获得阳性转化子的概率约为2.4%。
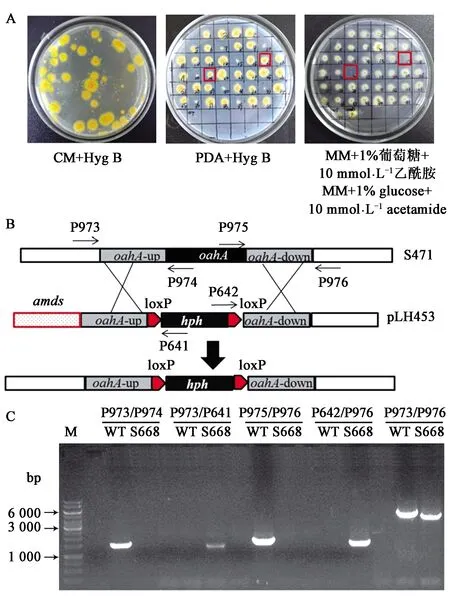
A:oahA基因敲除转化子在不同培养基上的生长情况,红框所示为敲除转化子;B:oahA基因敲除结构;C:黑曲霉ΔoahA菌株PCR结果,M:DNA分子质量标准;WT:黑曲霉野生型菌株S304;S668:oahA敲除转化子。
2.3 oahA回补菌株构建及分析
利用包含oahA表达盒的回补质粒pLH372转化无hph抗性筛选标记的ΔoahA菌株,质粒中的loxP-hph-loxP赋予宿主菌体在含有潮霉素B培养基上的生长能力(图3A)。为进一步将潮霉素筛选标记删除,将所得转化子孢子涂布于含有30 μg·mL-1四环素的MM平板上,诱导培养7 d后挑取转化子于PDA平板上,第2 d将长出的转化子对应点接至含有250 μg·mL-1潮霉素B的PDA抗性平板上,选择在PDA上生长而在潮霉素抗性平板上不生长的克隆,其中第24号转化子经PCR验证后为正确的oahA回补菌株,命名为S430(图3B)。同时,将黑曲霉野生型菌株S304,ΔoahA菌株S668和oahAΔoahA菌株S430接种于PDA培养基观察菌株的生长情况,发现三株菌的生长速度、产孢能力无显著差异(图3C)。
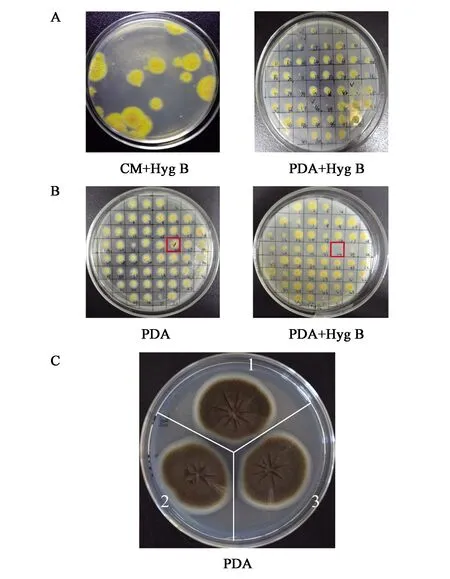
A:oahA回补S668生长情况;B:oahAΔoahA菌株S430生长情况,红框为回补菌株S430;C:三种菌株在PDA培养基上生产情况对比。
2.4 oahA基因对草酸合成的影响
2.4.1不同菌株中oahA转录及酶活水平的变化
为了考察oahA基因在黑曲霉发酵生产草酸中表达情况,S304、ΔoahA菌株S668和S430接种于黑曲霉草酸发酵培养基中培养3 d,收集菌丝体,提取总RNA后通过RT-PCR检测oahA基因在草酸发酵中的表达情况。如图4A所示,黑曲霉野生型菌株S304和oahAΔoahA菌株S430中,oahA基因均高水平表达,而ΔoahA菌株S668中无oahA基因表达。同时对三株菌中草酰乙酸水解酶的活性进行检测,发现与野生型菌株S304(0.16 U·mg-1)相比,ΔoahA菌株S668中未检测到草酰乙酸水解酶活性,而oahA回补菌株S430中草酰乙酸水解酶比酶活则恢复到野生型的水平(0.21 U·mg-1)(图4B)。
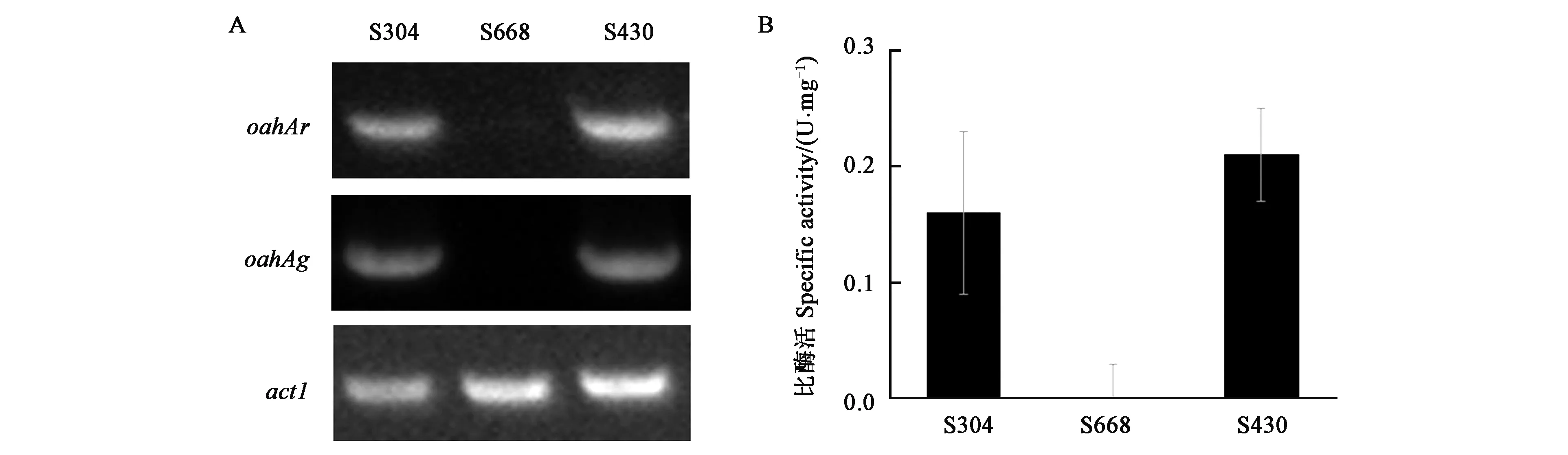
A:反转录PCR分析oahA在不同菌株中的转录情况;B:不同菌株中草酰乙酸水解酶酶活性测定。
2.4.2不同菌株合成草酸水平分析 为了考察oahA基因敲除后黑曲霉草酸产量的变化,通过HPLC方法对黑曲霉发酵液进行分析(图5A)。结果显示,黑曲霉野生型菌株S304草酸产量为10.3 g·L-1,而oahA基因敲除菌株S668为0.87 g·mL-1,草酸产量显著下降,回补oahA基因后,草酸产量恢复到野生型水平(图5B)。同时在此发酵过程中,三种菌体干重没有显著差异(图5C)。
3 讨论
黑曲霉作为一种重要的工业生产菌株在有机酸和酶制剂生产中得到了广泛应用,然而对其遗传学研究还不充分。由于可用于黑曲霉转化体系的遗传筛选标记有限,通过黑曲霉的遗传改造提升工业菌种生产性能步伐缓慢。构巢曲霉中的amdS基因编码一种乙酰胺酶,对于该基因的表达调控机制已做了详细研究[8-9],并且利用该基因先后在构巢曲霉[14]、玉米病原菌异旋孢腔菌[15]、产黄青霉菌[16]、纳地青霉[17]、米曲霉[18]和其他黑曲霉菌株[3,19]等多种丝状真菌中建立起遗传转化系统。在黑曲霉中并不存在amdS的同源基因。通过研究发现,野生型黑曲霉菌株在以乙酰胺为唯一氮源的培养基上生长不良,而amdS表达盒则赋予了其生长能力(图1)。这为在黑曲霉中构建以amdS为筛选标记的遗传转化体系提供了可能性。本文以构巢曲霉中的amdS基因表达盒构建了黑曲霉基因敲除出发质粒pLH439,当通过同源重组发生基因敲除时,因amdS表达盒被丢失而丧失在乙酰胺为唯一氮源培养基上的生长能力(图1和图2)。黑曲霉中草酰乙酸水解酶oahA是柠檬酸发酵生产中草酸的主要来源,通过构建的以amdS为筛选标记的基因敲除系统,成功地筛选到ΔoahA黑曲霉菌株。ΔoahA黑曲霉菌株菌体生长能力无显著变化(图3),而草酸合成水平显著降低(图5),由此证明以amdS为筛选标记的黑曲霉遗传转化体系成功建立,且在构建草酰乙酸水解酶敲除菌株过程中,利用该系统获得阳性转化子的概率为2.4%。amdS筛选标记的引入大大提高了筛选效率,减少了实验工作量。同时amdS为一种营养筛选标记,在食品、药品生产用菌株的改造中有显著优势,这为理性高效改造黑曲霉菌株提供了有力工具。
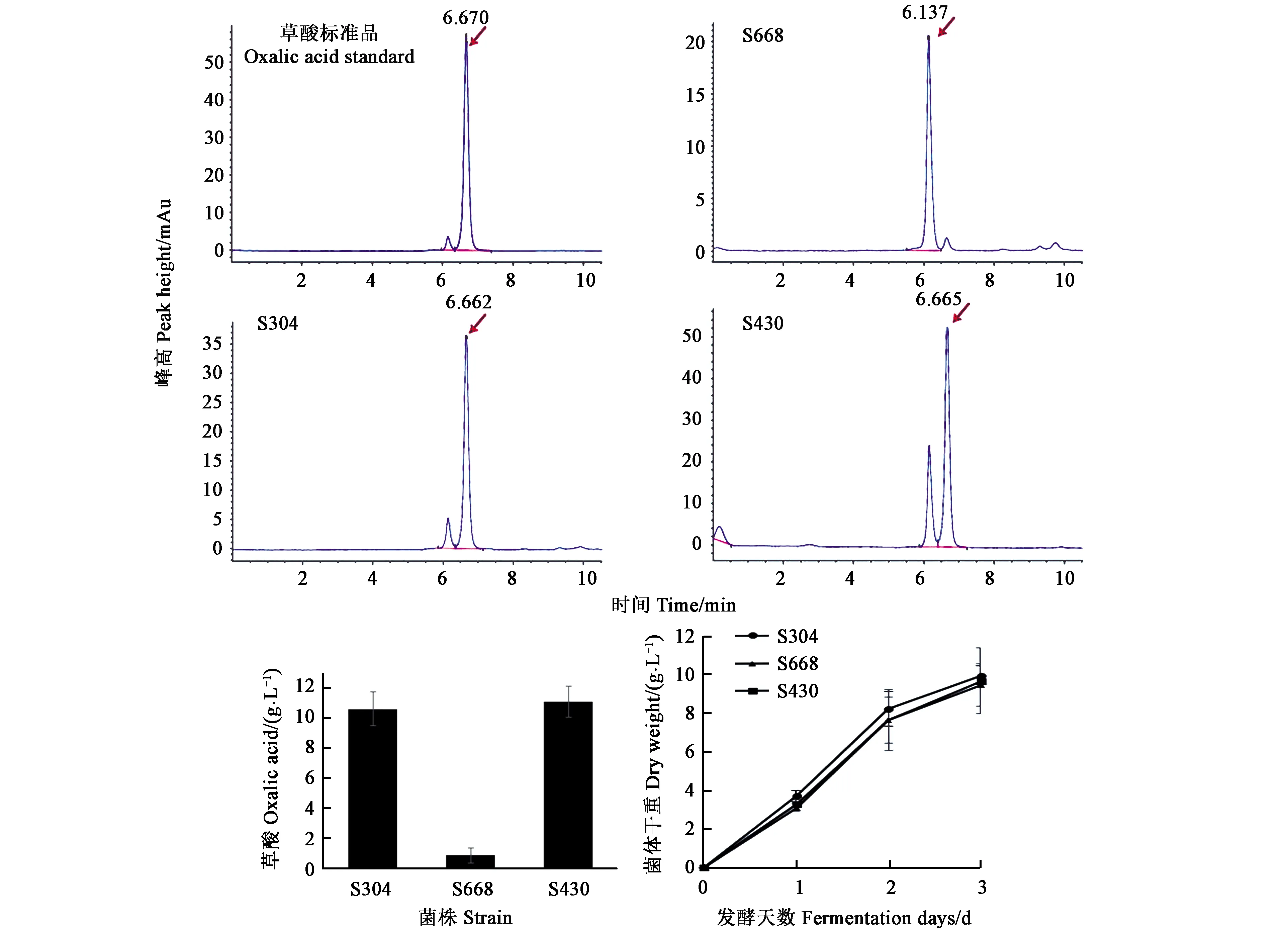
A:通过HPLC方法分析黑曲霉发酵液中草酸含量(红色箭头所指位置即为草酸,保留时间为6.6 min);B:不同菌株在草酸产量;C:发酵过程中菌体干重的变化。
