新型亲水性共轭聚合物的制备及光催化制氢性能
2020-03-12梁佑才胡志诚唐浩然刘孝诚邢晔彤
罗 威,梁佑才,胡志诚,唐浩然,刘孝诚,邢晔彤,黄 飞
(华南理工大学发光材料与国家重点实验室,广州 510640)
光催化分解水技术能够制备清洁、环保及高能量密度的氢燃料,为解决日益增长的能源需求问题提供了巨大的可能[1]. 自1972年Fujima等[2]采用二氧化钛实现光催化制氢以来,光催化半导体材料得到了广泛的研究与开发[3~7]. 共轭聚合物作为一种有机半导体已被广泛应用于有机光电器件(如有机电致发光及有机太阳能电池等)领域[8~11]. 共轭聚合物的结构可以通过化学反应调控,进而改变其吸收光谱、能级、载流子迁移率、孔隙率及光电性能等. 近年来,共轭聚合物在光催化制氢方面的应用备受关注[12~19]. 石墨氮化碳(g-C3N4)[20~31]、共价有机框架[32]和多孔/线性共轭聚合物[33~51]等多种共轭材料已被陆续开发出来,并且表现出了光催化制氢性能.
为了实现高效率的光催化制氢,共轭聚合物需要具备较宽的吸收光谱、合适的能级及在水中良好的分散性[12~19]. 通过将给电子单元和吸电子单元进行共聚,可制备吸收光谱及能级均可调的共轭聚合物. 此外,通过引入亲水侧链可以使共轭聚合物在水中具有良好的分散性[52~56]. 通常,共轭聚合物类的光催化制氢反应需要助催化剂(如铂)的协助[57],因此需要调控共轭聚合物与助催化剂之间的相互作用. 目前,大多数共轭聚合物的制氢效率相对较低,如何提高其光催化制氢效率是目前面临的挑战. 此外,关于共轭聚合物的化学结构和光催化性能之间的关系也尚未明确,需要进一步加强对共轭聚合物的结构-性能关系的研究. 含氮芳香环(如苯并噻二唑及吡啶等)是一类具有较大潜力的制备高效共轭聚合物光催化剂的构筑单元. 苯并噻二唑具有较强的吸电子能力,可用于构筑共轭聚合物用于光催化制氢[35,58~60]. 本课题组前期的研究结果[54]也表明,基于苯并噻二唑的共轭聚合物具有较低的氢结合自由能及较好的光催化性能. 目前用于构筑共轭聚合物类光催化剂的吸电子单元仍较少,需要进一步开发新型构筑单元,并研究其结构与催化剂性能之间的关系. 本文制备了3,6-二(噻吩-2-基)-1,2,4,5-四嗪(4N)和2,5-二(噻唑-2-基)-吡嗪(2N-2N)两种新型杂环,将其与亲水性单体进行共聚,制备了聚({4,8-双[(2,5,8,11,14,17,20-七氧二十二烷-22-基)氧基]苯并[1,2-b∶4,5-b′]二噻吩}-交替-[2,5-二(噻唑-2-基)吡嗪])(P7O-2N-2N)和聚({4,8-双[(2,5,8,11,14,17,20-七氧二十二烷-22-基)氧基]苯并[1,2-b∶4,5-b′]二噻吩}-交替-[3,6-双(5-溴-2-噻吩基)-1,2,4,5-四嗪])(P7O-4N)2种亲水性共轭聚合物光催化剂,其结构式如图 1所示. 采用密度泛函理论计算了含氮杂环氮位点的氢结合自由能(ΔGH),结果表明,P7O-4N中氮位点的ΔGH比P7O-2N-2N中氮位点的ΔGH更接近0; 表征了P7O-4N和P7O-2N-2N的紫外-可见吸收光谱、电化学特性及光催化制氢性能,研究结果表明,与P7O-2N-2N相比,P7O-4N具有更好的链间聚集特性、更红的吸收光谱及更高(6倍)的光催化制氢效率. 该研究结果对于光催化制氢的共轭聚合物的结构设计及性能提升具有参考意义.

Fig.1 Chemical structures of P7O-2N-2N(A) and P7O-4N(B)
1 实验部分
1.1 试剂与仪器
无水四氢呋喃、无水甲苯、氯化三丁基锡、亚硝酸异戊酯、液溴、硫粉、四(三苯基磷钯)三(二亚苄基丙酮)二钯和三(邻甲基苯基)磷,萨恩化学技术(上海)有限公司; 正丁基锂/正己烷,百灵威科技有限公司; 二氯甲烷、乙酸乙酯、甲醇、正己烷、水合肼、氯苯、N,N-二甲基甲酰胺、石油醚和乙醇,广州化学试剂公司; 无水硫酸镁、三氧化二铝、硅胶粉、碳酸钾和硫代硫酸钠,上海润捷化学试剂有限公司; 去离子水,广州亚飞水处理设备有限公司; 2-溴噻唑(1)、2-氰基噻吩(4)和2,5-二溴吡嗪,华威锐科化工有限公司; 所有试剂均为分析纯.
Bruker AV-500 MHz核磁共振波谱(1H NMR和13C NMR)仪,瑞士Bruker公司,溶剂为氘代氯仿; Waters 2410型高温液相色谱(GPC)仪,美国Waters公司,以CHCl3为测试流动相; HP 8453型紫外-可见(UV-Vis)分光光度仪,美国惠普公司,溶液吸收测试浓度为0.02 mmol/mL,制备薄膜吸收样品溶液浓度为10 mg/mL,溶剂为氯仿; CHI660E型电化学工作站,上海辰华仪器有限公司,以玻碳电极为工作电极,饱和甘汞电极为参比电极,铂丝为对电极,0.1 mol/L四丁基六氟磷酸胺(Bu4NPF6)的乙腈溶液为测试电解质; JEOL-2100F型高分辨透射电子显微镜(HRTEM),日本电子株式会社; Absolar-ⅢAG型光催化在线分析系统,北京泊菲莱科技有限公司; SK5200GT型超声仪,上海科导超声仪器有限公司; GC7900型气相色谱仪,上海天美科学仪器有限公司.
1.2 单体及聚合物的合成
将4.92 g(30 mmol)2-溴噻唑(1)加入到250 mL长颈两口烧瓶中,抽换气3次后加入50 mL无水四氢呋喃,搅拌均匀; 将烧瓶置于-78 ℃的低温浴中,在搅拌条件下滴加13.2 mL 2.5 mol/L的正丁基锂/正己烷溶液; 滴加完毕后,在-78 ℃低温浴中搅拌1 h,然后加入10.74 g(33 mol)氯化三丁基锡,继续反应1 h; 逐渐恢复到室温,搅拌16 h后加入30 mL去离子水淬灭反应. 反应体系经乙酸乙酯(60 mL×3)萃取和水洗(120 mL×3)后,用无水硫酸镁干燥,过滤并浓缩; 粗产物用三氧化二铝柱层析(正己烷作为洗脱剂)处理,得到2-(三丁基锡)-噻唑(2)黄色液体产物.
将3.74 g(10 mmol)化合物2和0.95 g(4 mmol)2,5-二溴吡嗪置于250 mL两口圆底烧瓶中,加入100 mL无水甲苯并搅拌均匀; 抽换气3次后,加入0.23 g(0.2 mmol)四(三苯基膦)钯,并通气15 min; 升温至100 ℃,反应6 h; 待反应温度降至室温后,用乙酸乙酯(60 mL×3)萃取并水洗(120 mL×3),用无水硫酸镁干燥,过滤; 所得溶液经旋转蒸发干溶剂后,用二氯甲烷/石油醚(体积比1∶3)作为洗脱剂,通过硅胶柱层析进行提纯,得到2,5-(二噻唑)-吡嗪(2N-2N,3)0.69 g,产率70%.1H NMR(500 MHz,CDCl3),δ: 9.42(s,2H),8.04~8.03(d,2H),7.57~7.56(d,2H);13C NMR(125 MHz,CDCl3),δ: 167.12,142.83,141.85,138.81,117.77.
将0.492 g(2 mmol)化合物3加入50 mL圆底烧瓶中,然后加入10 mL二氯甲烷,搅拌均匀; 向烧瓶中加入1.38 g(10 mmol)碳酸钾,并逐滴加入液溴1.58 g(10 mmol),室温下搅拌12 h; 加入10 mL 0.5 mol/L硫代硫酸钠淬灭反应后,加入二氯甲烷(30 mL×3)萃取,水洗(90 mL×3),用无水硫酸镁干燥后过滤; 所得溶液经浓缩后使用二氯甲烷/石油醚(体积比1∶2)作为洗脱剂通过硅胶柱层析提纯,得到单体2,5-[二(5-溴噻唑-2-基)]-吡嗪(M1)0.73 g,产率90%.1H NMR(500 MHz,CDCl3),δ: 9.29(s,2H),7.88(s,2H);13C NMR(125 MHz,TFA-d3),δ: 167.10,142.80,141.86,138.87,117.76.
将3.27 g(30 mmol)2-氰基噻吩(4)加入到干燥的圆底烧瓶中,并依次加入0.64 g(20 mol)硫粉、3.2 g(50%~60%,质量分数)水合肼和15 mL乙醇; 将反应液加热至80 ℃,反应2 h后冷却至室温; 通过抽滤收集棕色沉淀物3,6-二(噻吩-2-基)-1,2-二氢-1,2,4,5-四嗪(5),真空干燥; 化合物5未经纯化,直接用于下一步反应.
将化合物5置于250 mL圆底烧瓶中,加入亚硝酸钠(30 mmol,2.07 g)和醋酸(120 mL),室温下搅拌16 h; 通过旋转蒸发仪除去溶剂,粗产物采用二氯甲烷/石油醚(体积比1∶2)作为洗脱剂通过硅胶柱层析提纯,得到3,6-二(噻吩-2-基)-1,2,4,5-四嗪(4N,6)红色粉末1.58 g,产率43%.1H NMR(500 MHz,CDCl3),δ: 8.90~8.80(m,2H),7.88~7.70(m,2H),7.29~7.26(m,2H);13C NMR(125 MHz,CDCl3),δ: 161.5,136.0,132.5,131.0,129.0.
采用与合成M1相同的方法由化合物6合成3,6-二(5-溴噻吩-2-基)-1,2,4,5-四嗪(M2),得到红色粉末产物1.3 g,产率85%.1H NMR(500 MHz,CDCl3),δ: 8.02~8.01(d,2H),7.24~7.23(d,2H);13C NMR(125 MHz,CDCl3),δ: 160.71,137.04,132.13,131.31,120.99.
参照文献[54]方法制备{4,8-双[2-(2-甲氧基乙氧基)乙氧基]苯并[1,2-b∶4,5-b′]二噻吩-2,6-二基}双(三甲基锡烷)(M3). 将0.2 mmol M3和0.2 mmol M1加入25 mL圆底烧瓶中,再加入4 mL氯苯和2 mLN,N-二甲基甲酰胺,搅拌均匀; 抽换气3次除去氧气后,加入2 mg三(二亚苄基丙酮)二钯和4 mg三(邻甲基苯基)磷,再抽换气一次; 加热至110 ℃并反应4 h; 将反应后的溶液在甲醇中沉淀; 用氯仿溶解收集的沉淀,浓缩后再次在甲醇中沉淀,重复操作3次; 将得到的聚合物固体真空干燥,得到聚合物P7O-2N-2N 118 mg,产率85%.1H NMR(500 MHz,CDCl3),δ: 9.42~9.36(m,1H),8.13~8.19(m,1H),8.01~8.04(m,1H),7.54~7.59(m,1H),7.43~7.42(d,1H),3.36~4.49(m,66H). GPC(CHCl3),Mn=15600,分子量分布(D-)=1.63.
以0.2 mmol M2代替0.2 mmol M1,采用相同的方法合成聚合物P7O-4N,得到117 mg深紫色黏稠状固体,产率84%.1H NMR(500 MHz,CDCl3),δ: 8.25~7.74(m,4H),7.59~7.40(m,2H),3.35~4.49(m,66H). GPC(CHCl3),Mn=12700,D-=1.83.
1.3 光催化制氢实验
将2.5 mg聚合物溶解于250 μL四氢呋喃中,进一步分散在50 mL抗坏血酸水溶液(0.2 mol/L,用1 mol/L NaOH溶液调节溶液的pH=4)中,超声分散30 min后加入25 μL H2PtCl4溶液; 将反应液脱气30 min除去溶解氧和残留的少量四氢呋喃,然后采用氙灯(300 W,Ceaulight)光照60 min原位生成Pt纳米颗粒后开始光催化制氢实验. 反应液表面的光功率采用功率计校准为150 mW/cm2; 采用具有三电极池的CHI660E电化学系统测试光电流响应. 测试中采用Pt片作为对电极,Ag/AgCl电极作为参比电极,0.1 mol/L的Na2SO4水溶液(pH=6.8,除氧)作为电解质. 通过将聚合物的氯仿溶液(浓度为10 mg/L)涂覆到ITO导电玻璃上干燥成膜(厚度约为300 nm)制备工作电极. 光电流响应测试样品的有效面积为0.50 cm2.
2 结果与讨论
2.1 合成与表征
P7O-2N-2N和P7O-4N的合成路线如Scheme 1所示. P7O-2N-2N和P7O-4N分别由单体M1,M2与M3采用Still聚合反应得到. 利用1H NMR确定P7O-2N-2N和P7O-4N的化学结构,其平均分子质量通过GPC(氯仿作为流动相)测定. GPC结果如表1所示,P7O-2N-2N和P7O-4N的数均分子量分别为15600和12700,分子量分布分别为1.63和1.83.

Scheme 1 Synthetic routes of 2,5-bis(5-bromothiazol-2-yl)pyrazine(2N-2N,3), 3,6-bis(5-bromothiophen-2-yl)-1,2,4,5-tetrazine)(4N,6), P7O-2N-2N and P7O-4N

Table 1 Molecular weights,optical properties,and energy levels of P7O-2N-2N and P7O-4N
2.2 密度泛函理论计算
为了研究P7O-2N-2N和P7O-4N的化学结构与光催化性能之间的关系,采用基于密度泛函理论(DFT)的计算方法对聚合物的氢结合自由能(ΔGH)进行了模拟计算. ΔGH可以作为水还原反应效率高低的一个判断依据[39,61~63]. ΔGH值越大,氢原子越难被催化位点吸附,越容易发生质子化反应; ΔGH值越小,氢原子越容易被活性位点吸附,同时会对氢气的产生及释放产后阻碍. 通常,ΔGH接近于0是实现水还原反应的理想值. 在含氮杂环的聚合物中,氮原子被认为是制氢过程中的催化活性位点[39]. 因此,本文计算了P7O-2N-2N和P7O-4N的含氮杂环[2N-2N和4N,图2(A)]中氮原子的ΔGH值. 由图2(B)可以看出,2N-2N和4N中氮杂环上α位点氮原子的ΔGH值分别为0.746和0.061 eV,β位点氮原子的ΔGH值分别为0.600和0.005 eV. 含氮杂环4N中α氮原子和β氮原子的ΔGH值均比2N-2N中的更接近于0,因此初步推断P7O-4N的光催化制氢活性要强于P7O-2N-2N.

Fig.2 Chemical structures of 2N-2N and 4N(A) and ΔGH values of nitrogen site in 2N-2N and 4N(B)
2.3 UV-Vis吸收光谱
图3为P7O-2N-2N和P7O-4N在氯仿溶液和薄膜状态下的UV-Vis吸收光谱. 由图3可见,由于聚合物中氮原子的位置不同,两种聚合物的吸收图谱也表现出一定的差异. P7O-2N-2N溶液的吸收光谱在510 nm处出现了一个吸收峰,归属于P7O-2N-2N主链的分子内电荷转移; P7O-4N在533和570 nm处出现2个吸收峰,其中570 nm处的肩峰来自聚合物主链上较强的π-π相互作用,表明P7O-4N的链间聚集作用比P7O-2N-2N的链间聚集作用更强,有利于电荷传输. 两个聚合物的薄膜吸收光谱相对于溶液的吸收图谱都有略微的红移. P7O-2N-2N的薄膜吸收边为629 nm,P7O-4N的吸收边为655 nm. 通过公式Eopt=1240/λedge算出P7O-2N-2N和P7O-4N的光学带隙(Eopt,eV)分别为1.97和1.89 eV.

Fig.3 UV-Vis absorption spectra of P7O-2N-2N(a) and P7O-4N(b) in chloroform(A) and thin film(B)
2.4 电化学特性和电子能级结构
采用循环伏安(CV)法测定聚合物的氧化还原电位. 以二茂铁的氧化电位作为标定电位,Bu4NPF6的乙腈溶液作为测试电解质溶液. 采用下式计算聚合物的最高占有轨道能级(EHOMO,eV)和最低未占有轨道能级(ELUMO,eV):
EHOMO=-e(Eox+4.53)
(1)
ELUMO=-e(Ered+4.53)
(2)
式中:Eox(V)和Ered(V)分别为聚合物的氧化和还原电位. 由图4(A)可以看出,P7O-2N-2N和P7O-4N的氧化电位分别为0.94和0.76 V,还原电位分别为-0.66和-0.71 V. 通过计算得出,P7O-2N-2N的EHOMO为-5.47 eV(vs.Ev,真空能级),ELUMO为-3.87 eV; P7O-4N的EHOMO为-5.29 eV,ELUMO为-3.82 eV. 图4(B)为聚合物P7O-2N-2N和P7O-4N的能级图,其中虚线表示标准氢电极在溶液pH=4时的还原电势(-4.23 V)[51]. 可以看出,两种聚合物的ELUMO均高于水的还原电势,说明两种聚合物都具备较大的还原水的驱动力[63].
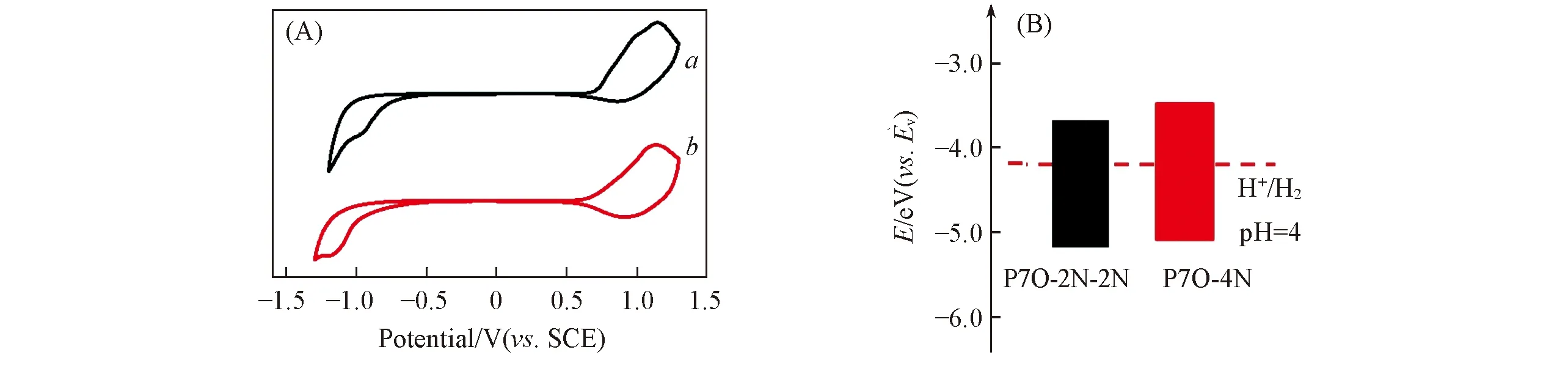
Fig.4 CV curves(A) and energy level alignment diagram(B) of P7O-2N-2N(a) and P7O-4N(b)
2.5 聚合物的透射电子显微镜表征
由于P7O-2N-2N和P7O-4N的侧链引入了寡聚乙二醇侧链,聚合物在水中具有良好的分散性. 共轭聚合物在水中较好的分散性能够增加聚合物和水的界面接触面积,有利于提高光催化制氢性能[51~53,64,65]. 图5为聚合物分散在水中的TEM照片. 可以看出,P7O-2N-2N和P7O-4N都可以很好地分散在水中,并形成尺寸为几十纳米的无定形聚集体. 更小尺寸的聚集体有利于助催化剂在聚合物表面的负载,增大负载面积并缩短激子解离后迁移到聚合物/助催化剂界面的距离,有助于增强光催化制氢性能.

Fig.5 TEM images of P7O-2N-2N(A) and P7O-4N(B) dispersed in waterInsets: enlarged TEM images of the corresponding polymer.
2.6 聚合物的光催化制氢性能

Fig.6 Photocatalytic hydrogen evolution rates of P7O-4N(a,b) and P7O-2N-2N(c,d) with Pt(a,c) and without Pt(b,d)
图6为聚合物溶液加入Pt助催化剂前后光催化制氢量随光照时间的变化图. 可以看出,2种聚合物溶液在加入Pt助催化剂时的氢气产生速率要显著高于未加入Pt助催化剂时的氢气产生速率; 无论是否加入Pt助催化剂,聚合物P7O-4N均呈现出比P7O-2N-2N更强的光催化制氢性能. 计算结果表明,在不加助催化剂时,P7O-4N反应液的氢气产生速率为0.142 μmol/h(0.057 mmol·g-1·h-1),P7O-2N-2N反应液的氢气产生速率为0.033 μmol/h(0.013 mmol·g-1·h-1). 在加入Pt纳米颗粒时,P7O-4N的氢气产生速率为3.9 μmol/h(1.56 mmol·g-1·h-1),而P7O-2N-2N的氢气产生速率只有0.6 μmol/h(0.24 mmol·g-1·h-1),P7O-4N的氢气产生速率是P7O-2N-2N的6.5倍. 这一结果与DFT模拟计算结果一致. 与P7O-2N-2N相比, P7O-4N的链间聚集特性更强,吸收光谱更加红移,有利于其光催化制氢性能的提升.
2.7 聚合物的光电流响应
为了进一步探究2个聚合物光催化性能的差异,测试了聚合物的薄膜的光电流响应. 图7为聚合物在外加电压为0或-0.2 V下的光电流响应测试结果. 当电极上的外加电压为0时,P7O-4N的电流响应强度为5.1 μA/cm2,P7O-2N-2N的光电流响应强度为1.0 μA/cm2. 可以看到,与P7O-2N-2N样品相比,P7O-4N样品表现出更高电流密度响应. 当外加电压为-0.2 V时也出现了同样的现象. 与P7O-2N-2N相比,P7O-4N内部的光生载流子更容易解离[46],与光催化制氢结果一致.
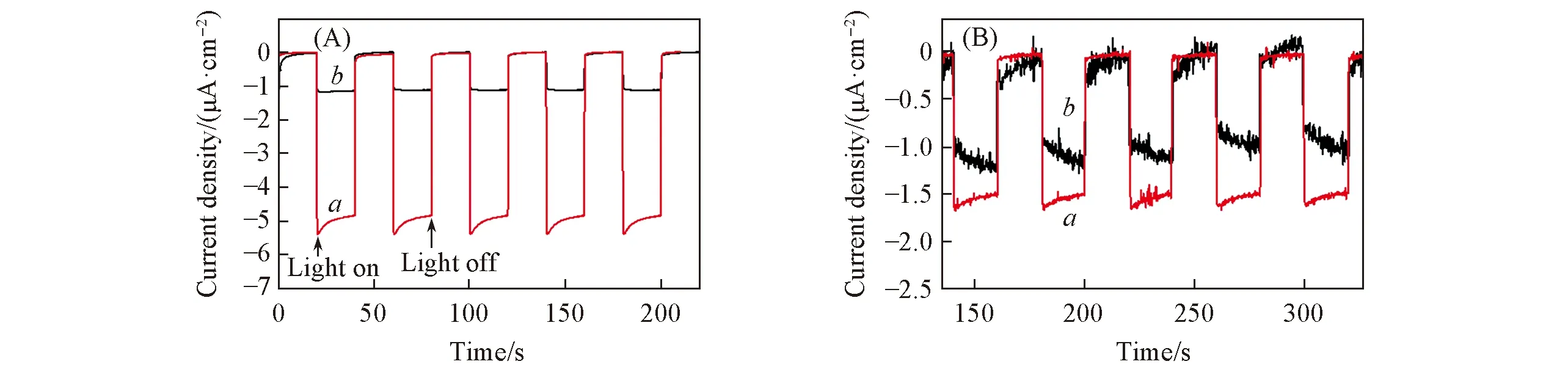
Fig.7 Photocurrent response of P7O-4N(a) and P7O-2N-2N(b) at applied voltages of 0 V(A) and -0.2 V(B)
3 结 论
设计并合成了2种具有不同含氮杂环受体单元的聚合物P7O-2N-2N和P7O-4N. P7O-2N-2N和P7O-4N的寡聚乙二醇侧链使其在水中具有良好的分散性. P7O-4N主链3,6-二(噻吩-2-基)-1,2,4,5-四嗪单元的ΔGH相比于P7O-2N-2N主链的2,5-二(噻唑-2-基)-吡嗪单元更接近于0. 与P7O-2N-2N相比,聚合物P7O-4N链间聚集特性更强,吸收光谱更加红移. 光催化研究结果表明,P7O-4N表现出比P7O-2N-2N更好(6倍)的光催化制氢性能. 该研究结果可为用于新型共轭聚合物类光催化剂的设计提供参考.
