利用三代测序技术识别小鼠早期胚胎等位基因特异的转录本和剪切事件
2020-03-10袁杰李敏黄诗圣舒文杰任超
袁杰,李敏,黄诗圣,舒文杰,任超
1.军事医学研究院 辐射医学研究所,北京 100850;2.广州大学 生命科学学院精准基因编辑工程中心,广东 广州 510006;3.上海科技大学 生命科学与技术学院,上海 201210
尽管高通量的短读长测序极大地促进了转录组学研究[1],然而基于组装的短读长测序不足以精确地进行生物信息学分析[2]。此外,先前研究表明真核生物的转录组非常复杂。包括可变剪切在内的前体mRNA的转录后加工极大地增加了转录组的多样性[3-4]。由于高通量测序的长度限制,它无法完全解析真核生物的转录组,特别是与真核生物中普遍存在的新型可变剪切事件相关联的复杂性[5-6]。近年出现的第三代测序技术,即长读长测序,通过使用长读长技术来实现碱基序列的实时读取,缩短了测序时间。此外,可以通过直接获得错误率小于1%的全长转录本来克服二代短读长转录组测序技术(RNA-seq)的测序长度限制[7]。最近,长读长测序被用来证明,即使是来自人体器官的高度表征过的转录组在基因和同源异构体层面也是不完整的[8-9]。相比之下,使用PacBio单分子实时(single-molecule real-time,SMRT)测序对玉米和草莓的转录组进行分析,发现了许多新颖的剪切同源异构体、长链非编码RNA(long noncoding RNAs,LncRNA)、融合转录本,以及新颖的可变剪切事件[10-11]。
等位基因特异性表达(allele-specific expression,ASE)是指二倍体生物体中来自2个等位基因的转录本的相对表达水平。等位基因特异性表达可能是由于转录速率、mRNA稳定性或其他影响转录本丰度的机制的不同所造成的[12]。在小鼠早期胚胎发育过程中,它会经历大规模重编程过程,以完成母源mRNA的降解和合子基因组激活(zygote genome activation,ZGA)[13]。这些重编程过程可以帮助调节胚胎基因组转录的激活,并为随后的胚胎发育和分化奠定基础[14]。
早期胚胎的起源效应分析可以鉴定出某些显示出ASE的基因,从而大大增强了我们对早期胚胎发育过程中重编程的理解。先前的研究表明,包括等位基因特异的剪切和甲基化不对称性在内的转录组和表观组的起源效应极大地影响着早期胚胎的发育[15]。然而,使用二代短读长测序技术从2个等位基因的相对丰度推断转录本来源的方法具有较大的局限性。在最近的研究中,我们利用三代测序技术生成了包括新颖转录本在内的更加完整的转录组[16]。在本研究中,我们应用三代测序技术描绘了小鼠早期胚胎的剪切图谱和ASE。我们还将三代与二代RNA-seq数据结合在一起,以获得完整的剪切图谱,并且利用更完整的剪切信息探究起源效应。我们的目标是更好地表征剪切图谱和转录组的等位基因特异性的注释信息,以增强对小鼠胚胎发育的了解。
1 材料和方法
1.1 材料
本研究用到的小鼠早期胚胎的二代测序数据、三代测序数据、全长转录本及三代长读长测序数据识别的可变剪切事件和差异可变剪切事件均来自GSE138760。
1.2 RNA-seq数据预处理
首先用 TrimGalore(v0.6.1)修剪 RNA-seq数据,随后用STAR(v2.5.0a)[17]将修剪后的数据比对到mm10参考基因组(参数:--two pass Mode Basic-outSAM typeBAM Unsorted-outSAM strand Field intron Motif)。用 Cufflinks(v2.2.1)[18]拼接各阶段的短读长转录本,并且过滤掉不包含正负链信息和每千个碱基的转录每百万映射读取的片段(fragments per kilobase million,FPKM)的值小于1的转录本。
1.3 等位基因特异性识别
为了将杂交小鼠(♂ DBA/2×♀C57/BL6J)的序列无偏映射到参考基因组,通过SNPsplit(v0.3.4),采用来自桑格小鼠基因组计划数据库(dbSNP142)的DBA/2特异的单核苷酸多态性(single nucleotide polymorphisms,SNPs)生成一套伪基因组。随后,用GMAP[19]将高质量的三代长读长转录本比对到N掩盖(N-masked)的基因组,并使用STAR将二代RNA-seq序列比对到N-masked的基因组。根据在N碱基处的等位基因特异性错配的SNP数量判定三代长读长转录本的来源。基于N位点处的碱基,使用SNPsplit将唯一比对上(uniquely mapped)的二代RNA-seq序列分为DBA/2特异组、C57/BL6J特异组和不可分配组。
为了识别等位基因特异的二代短读长转录本,首先过滤掉在任一生物重复样本中C57特异序列(C57 reads)+DBA特异序列(DBA reads)小于5的转录本。等位基因比率计算公式为ReadsC57/(ReadsC57+ReadsDBA)。然后,应用卡方检验计算每个重复样本中等位基因特异的序列偏性的P值,并且用fisher方法将P值合并。依据等位基因比率和P值来定义等位基因平衡和等位基因不平衡的转录本。│等位基因比率-0.5│<0.16或P≥0.05的转录本被定义为等位基因平衡的转录本,│等位基因比率-0.5│≥0.16且P<0.05的转录本被定义为等位基因不平衡的转录本。
采用2种不同的分析策略,比较三代测序数据与二代测序数据中的等位基因特异性。长读长转录本的起源与等位基因特异的SNP一致,而短读长转录本的等位基因特异性是由等位基因的比率和偏性决定的。
1.4 计算等位基因特异的基因与同源异构体之间表达模式的相关性
通过 StringTie(v1.3.3b)[20],使用二代 RNA-seq数据计算长读长基因和同源异构体的总表达量(FPKM)和等位基因特异的表达量(FPKM)。计算等位基因特异的表达量时,输入的数据是等位基因特异的序列。用皮尔逊相关性检验计算基因和同源异构体的总表达量以及等位基因特异表达量之间的相关性,相关性系数(cor)>0且P<0.05的模式被定义为一致,其余模式被定义为不一致。
1.5 基因本体(gene ontology,GO)功能富集分析
使用 PANTHER(v14)[21],对新发现的等位基因特异的转录本和相关性模式为不一致的基因进行GO功能富集分析。
1.6 识别可变剪切事件与差异可变剪切事件
使用SUPPA2(v2.2.1)[22],在7个阶段识别7种类型的可变剪切事件,包括外显子跳跃(skipping exons,SE)、可 变 的 5′端(alternative 5′splice sites,A5)、可变的3′端(alternative 3′splice sites,A3)、内含子保留(retained introns,RI)、互斥外显子(mutually exclusive exons,MX)、可变起始外显子(alternative first exons,AF)、可变末端外显子(alternative last exons,AL)。由注释的转录本生成的事件被定义为注释事件,其余事件被定义为新颖事件。差异可变剪切事件是从我们最近发表的研究中获取的[16]。我们通过STAR使用二代数据计算比对到剪切结上的序列(reads)。与等位基因特异的转录本相关联的可变剪切事件或差异可变剪切事件被定义为等位基因特异的可变剪切事件或差异可变剪切事件。
1.7 可变剪切事件的累积数量分析
为了评估三代测序数据识别可变剪切事件的潜力,比较了通过二代数据和三代与二代数据的组合来识别可变剪切事件的累积数量。首先随机选取一个阶段的二代数据识别的转录本,接着使用Cuffmerge(v2.2.1)[18]合并单个阶段的转录本与前面所有阶段的转录本,随后用SUPPA2从合并的转录本中识别可变剪切事件,当7个阶段都被合并时,完成一次循环。将上述过程重复100次,在每个点计算可变剪切事件数量的平均值和99%置信区间。当我们对三代数据与二代数据的组合进行累积数分析时,与上述策略唯一的区别在于分析之前每个阶段的长读长与短读长转录本都已经被合并。
2 结果
2.1 使用二代测序数据和三代测序数据识别等位基因特异的转录本
为了表征PacBio三代测序技术在研究早期胚胎转录组中等位基因特异的转录本和可变剪切事件的优势,我们从我们最近发表的研究中收集了小鼠早期胚胎7个阶段[精子(sperm,SP)、卵母细胞(oocyte,Oo)、1细胞(1-cell,1C)、2细胞(2-cell,2C)、4细胞(4-cell,4C)、8细胞(8-cell,8C)和囊胚(blastocyst,BL)]的测序数据和注释数据。这些数据包括二代测序数据、三代测序数据、来自7个阶段的全长转录本,以及7个阶段合并的全长转录本。这些数据被用来鉴别等位基因特异的转录本、可变剪切事件和转录组的起源效应(图1A)。借助三代测序技术的优势,可以将等位基因特异的单核苷酸多态性SNP精确定位在单个转录本中,从而有助于准确识别转录本的来源。绝大多数(97%)的长读长转录本中至少存在1个等位基因特异的SNP,接近一半的转录本(46%)至少包含3个等位基因特异的SNP。因此,我们把至少包含3个等位基因特异的SNP的转录本定义为等位基因特异的转录本。然后,我们基于每个阶段获得的长读长转录本中存在的等位基因特异的SNP来确定长读长转录本的起源。这些转录本被分为可以区分来源的转录本[包括C57特异(母源)、DBA特异(父源)以及双等位基因的转录本]和无法区分来源的转录本(图1B)。与先前研究结果一致[13],从1细胞到囊胚阶段,C57特异的转录本所占比例逐渐上升,DBA特异的转录本逐渐下降(图1B)。在7个阶段中,我们鉴别出734~1288个等位基因特异的转录本(图1C)。在卵母细胞和精子期,我们观察到大量的C57特异或DBA特异的转录本,然而随着胚胎发育,双等位基因的转录本比例逐渐上升(图1C)。通过与GENCODE(vM20)注释进行比较,每个阶段平均鉴定出532个C57特异的转录本和397个DBA特异的转录本,参考我们先前的发现,每个阶段平均存在650个注释的转录本和234个新基因和同源异构体(图1D)[16]。
接着我们比较了三代测序数据和二代测序数据鉴别的等位基因特异的转录本。尽管由于测序深度的原因,三代数据中识别的等位基因特异的转录本要少于二代数据,但不管是对于三代数据(图1B、C)还是二代数据,等位基因特异的转录本的数量和比例都是从1细胞到囊胚阶段逐渐减少。然而两者之间的重叠率逐渐降低,在囊胚阶段两者识别等位基因特异的转录本仅6%是一致的(图1E)。在7个阶段中,仅被三代数据所识别的新发现的等位基因特异的转录本数量范围为378~872(图1F)。我们对这些新发现的等位基因特异的转录本做了GO分析,发现这些转录本与细胞代谢过程、感觉知觉和细胞周期过程存在关联。
我们进一步表征了从三代数据中鉴别出的新发现的等位基因特异的转录本,将其分为3种类型,即没有特异的二代数据支持(未被等位基因特异的短读长序列所识别)、等位基因特异的偏性相反(与三代数据识别的结果相比,二代数据识别结果的亲代定位是相反的)、双等位基因(存在等位基因特异的SNP但等位基因的表达水平无差异)(图1F)。例如,在囊胚阶段识别的基因PB.2249的2个异构体在三代数据中被识别为品系特异,但在二代数据中被错误地鉴定为双等位基因(图1G)。这些结果证明了使用三代数据鉴别等位基因特异的转录本的优势。
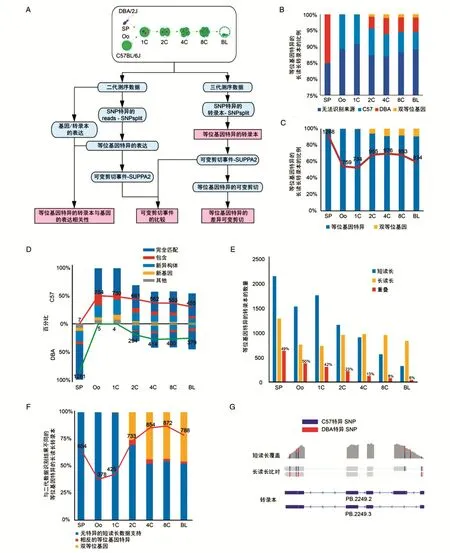
图1 三代测序数据与二代测序数据定义的等位基因特异的转录本
2.2 比较三代测序和二代测序数据中的可变剪切事件
为了表征三代数据识别可变剪切事件的优势,分别使用三代数据和二代数据在7个阶段识别可变剪切事件。基于GENCODE注释,这些事件被分为注释的事件和新颖的事件(图2A、B)。与二代数据相比,三代数据能鉴别出更多罕见的可变剪切事件,例如AF(P=1.3e-18,卡方检验),从而证明了三代测序技术捕获复杂可变剪切事件的能力。此外,从三代数据中识别的新颖事件的比例要显著大于二代数据中识别的新颖事件的比例(图2C)。这些结果证明,三代测序技术对于分析可变剪切事件极具价值。
接着,比较了分别从长读长转录本和短读长转录本中提取的剪切结(splicing junction,SJ)。大多数剪切结在两者之中都存在,但仍有数千个剪切结(平均每个阶段7058个)只能被三代数据所识别(图2D)。在这些仅能被三代数据所识别的特有的剪切结中,平均每个阶段有87%的剪切结能被至少5个短读长序列所支持(图2E)。这些结果表明借助三代测序技术,我们能识别出大量高准确度且特异的剪切结。
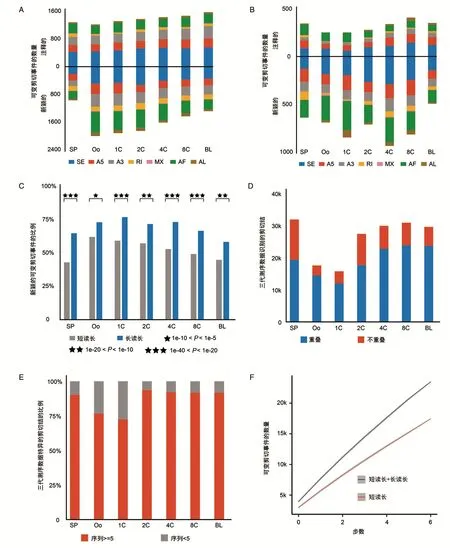
图2 使用三代测序数据和二代测序数据识别可变剪切事件
为了评估二代数据与三代数据的组合识别可变剪切事件的能力,我们比较了二代数据和二代与三代的组合数据识别可变剪切事件的累积数量。在当前的测序深度之下,从组合数据中识别出的事件显著地比仅从短读数据中识别的事件的数量多(P<1e-100,威尔逊配对秩和检验)(图2F)。这个结果显示出利用二代数据和三代数据的组合识别可变剪切事件的优势。
2.3 小鼠早期胚胎中与起源效应相关的同源异构体的表达
由于早期胚胎中亲本转录本的动态变化,可变剪切事件与等位基因特异的转录本存在关联。为了研究特定于起源的剪切异构体,我们对具有不少于2个转录本的基因以及关联转录本的总表达和等位基因特异的表达进行了定量。通过计算基因与同源异构体表达量之间的相关性,发现大多数异构体(73%)的表达模式与对应的基因一致。相反,有27%的异构体与基因表达模式不一致(图3A)。基于异构体与异构体等位基因特异性表达的相关性,我们将后者分为4类。我们观察到大多数异构体的表达模式与它们在等位基因特异性水平的表达不一致(图3B、C)。例如,Hsd17b6基因的C57特异性表达与一个新鉴定的Hsd17b6的异构体的表达一致,但是与其在整个基因层面的表达模式不一致;同样,Trim43a基因的DBA特异性表达与异构体的表达和其在整个基因层面的表达模式都不一致;然而,在ZGA过程中,Mcph1基因的异构体的DBA特异性表达与C57特异性表达模式相反(图3D、F)。我们对4组表达模式不一致的异构体进行了GO分析,这些基因参与了细胞代谢、基因表达及RNA加工等过程(图3G)。因此,我们可以通过整合三代测序数据和二代测序数据来识别阶段特异和等位基因特异的转录本。
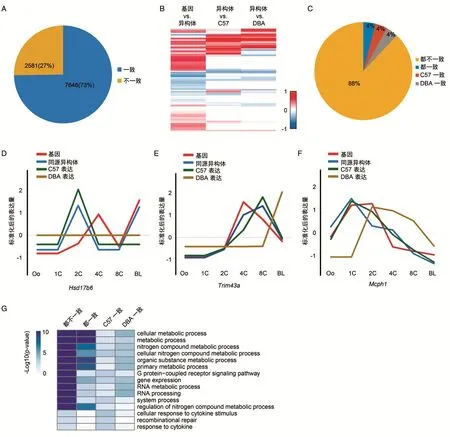
图3 等位基因特异的基因与转录本的表达模式的相关性
2.4 早期胚胎中基于三代测序数据的等位基因特异的可变剪切的分析
根据上述结果(图1、2),我们鉴定了一些新发现的等位基因特异的转录本,这些转录本可能是由等位基因特异的可变剪切事件产生的。因此,我们在三代测序数据中分析了等位基因特异的剪切事件的发生。每个阶段平均鉴定出230个等位基因特异的剪切事件(图4A)。我们观察到一些包含DBA特异与C57特异的mRNA的剪切事件。例如,在2细胞阶段,Tor1aip1基因的一个外显子跳跃事件产生了DBA特异的转录本TCONS_00001249和C57特异的转录本TCONS_00001250(无跳跃外显子)(图4B)。此外,我们结合三代和二代数据来检测早期胚胎发育过程中的等位特异的差异可变剪切事件(图4C)。等位基因特异的差异剪切事件在1细胞到2细胞阶段以及4细胞到8细胞阶段发生的频率更高。仍以Tor1aip1为例,在1细胞阶段,仅仅只有C57特异的转录本TCONS_00001250存在表达,然而DBA特异的转录本TCONS_00001249在1细胞到2细胞的转变中被激活(图4D、E)。随着一个新发现的DBA特异的差异可变剪切事件的出现(图4D、E),Tor1aip1基因中的DBA特异的转录本和C57特异的转录本的表达量都有所上升(图4F~H)。总的来说,这些数据表明某些亲本特异的转录本表现为亲本特异的剪切异构体,这些等位基因特异的剪切事件和差异可变剪切事件的发现将改善我们对早期胚胎发育过程的理解。
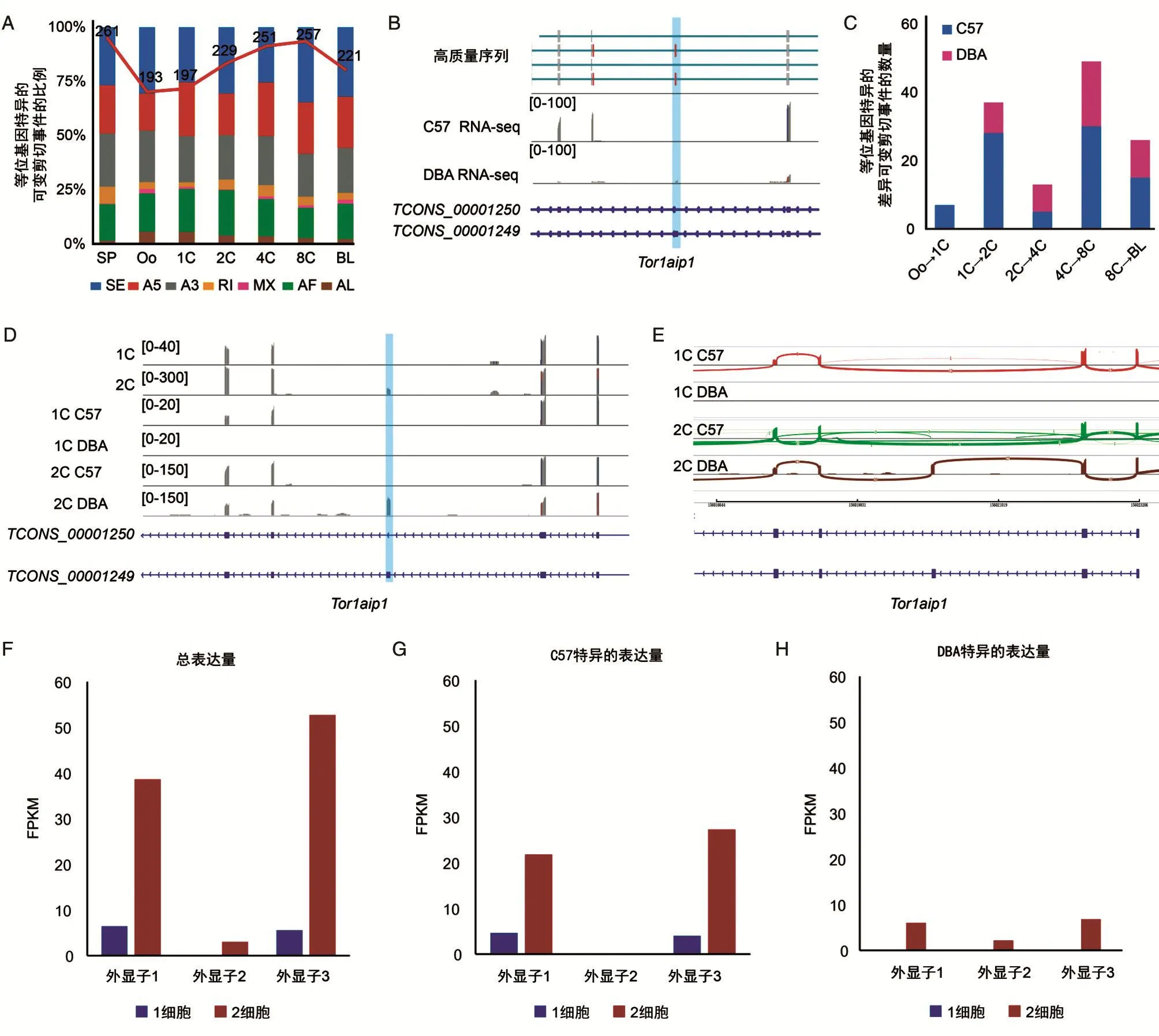
图4 基于三代测序数据的等位基因特异的可变剪切
3 讨论
在本研究中,我们使用了最近发表的研究中的高分辨率的转录组信息来分析起源效应,并且比较了小鼠早期胚胎的三代测序数据和二代测序数据识别的剪切事件[16]。我们的目的是探索三代测序技术在研究转录组起源效应方面的优势,并且鉴定新颖的可变剪切事件。
由于包含2个长距离的SNP的片段很难被扩增以及无法使用Sanger在整个序列中对800 bp以上的片段进行测序等技术限制,目前暂时无法对三代测序技术新识别的等位基因特异的转录本进行准确的实验验证。我们建立了一套生物信息学的流程来讨论三代测序技术在识别等位基因特异性方面的优势,并且借助这一优势来识别新的等位基因特异的转录本,在同源异构体的层面上研究了早期胚胎中等位基因特异的转录激活。结果显示,与基于组装的二代测序技术相比,三代测序技术能够更准确地识别等位基因特异的转录本和剪切事件。尽管二代数据能识别大量等位基因特异的转录本,但是考虑到这种基于组装的策略的可靠性与准确性的不足,我们仅关注于利用三代数据的准确性和优势来发现等位基因特异的转录本。随着母源mRNA在ZGA过程中降解[23],我们观察到在1细胞到2细胞转变过程中C57特异的转录本数量明显下降。但是即便是到囊胚期,仍有数百个转录本保持着等位基因失衡的状态,这表明等位基因特异的转录本是胚胎发育过程中的常规产物[24]。
三代测序数据具有极高的识别新颖可变剪切事件的潜力。我们对可变剪切事件的累积数量分析表明,将三代数据和二代数据合并可以快速增加可变剪切事件的数量。此外,三代数据有助于识别大量二代数据无法识别的新颖的剪切结。这突显了三代数据在识别新的剪切事件方面的优势。
我们还证明了,很大一部分基因的表达模式与它对应的异构体或它在亲本水平的表达都不一致。我们推断某些DBA或C57偏性的转录本在特定阶段被特异性激活,它们可能在功能上参与了胚胎发育过程。因此,迫切需要开发适当和有效的工具来阐明等位基因特异的转录本的功能[25-26]。最近的一项研究通过靶向非甲基化基因座实现了印记基因的等位基因特异性编辑[26],这可以用于进一步研究等位基因特异性基因在发育过程中的功能。
当依据等位基因特异的SNP来分离新颖的剪切异构体时,我们观察到数百个品系特异的剪切事件。总体而言,与DBA特异的转录本相比,识别出更多的C57特异的转录本,尤其是在1细胞和2细胞阶段。这些发现与已有的母源基因编码的转录因子激活了ZGA过程的推测一致[24]。在启动子区,基因间区和远端区域的等位基因特异的甲基化和母源组蛋白H3第27位赖氨酸三甲基化(H3 lysine 27 trimethylation,H3K27me3)修饰[27-29],在等位基因特异的可变剪切的分子调节过程中成为不依赖于DNA甲基化的印记机理[30-32]。这一过程可能控制了等位基因特异的基因表达和可变剪切事件。因此,利用三代测序技术识别父母源特异的DNA甲基化过程或H3K27me3印记现象,有助于进一步解释等位基因特异的转录调控。
总之,我们建议借助三代测序技术的优势准确识别等位基因特异的转录本与剪切事件。通过三代测序技术,我们可以获得更多等位基因特异的转录本、新颖可变剪切事件以及更多的剪切结,并且我们还报道了早期胚胎过程中的等位基因特异的可变剪切和差异可变剪切。这些发现可以加深我们对于早期胚胎发育过程中转录组起源效应的理解。
