生物矿化及仿生矿化中的信息传递和转化
2020-03-10潘海华唐睿康
潘海华,唐睿康
(1 浙江大学求是高等研究院,浙江杭州310027;2浙江大学化学系,浙江杭州310027;3浙江大学生物物质与信息调控研究中心,浙江杭州310027)
引 言
在当前科技飞速发展的同时,人类也面临人口爆炸、物质需求倍增与资源日益短缺的全球性难题,对化工生产的材料制备工艺也提出了更高的要求:高性能、绿色和可持续发展已成为全球的共识[1]。具有超级结构的有序复合材料相比单组分体相材料而言,具有用量少、性能高、多用途和智能性等优点,是材料化工制备发展的重要方向。当前,化工生产已从大宗量的通用单组分体相材料制备向多组分、多功能、个性化定制的复合材料的精细制备转变。超级结构有序复合材料的制备要求不同材料在微纳尺度以特定有序的方式装配在一起,这对传统化工的产品工程和过程控制提出了更高的挑战。但在自然界,生物体系经过亿万年的演化,已发展出一种高效、绿色的分子工程策略可制备出牙齿、骨骼和贝壳等具有多级有序的结构、优异的力学性能、良好生物相容性的生物矿物。该生物策略就是通过生物矿化的过程来调控纳米矿物的结晶和装配[2]。生物矿化过程是通过生物分子与矿物间的识别,将生物分子结构信息传递到矿物,并实现分子结构信息向矿物结构的转化,这与金涌院士[3]前瞻性提出的化学工程中物质、能量和信息的“三传三转”范式中的“信息传递和转化”,以及胡英院士[4]倡导的重视化学工程的分子工程发展方向不谋而合。本文将从生物矿物-溶液界面结构、生物分子与矿物晶面的分子识别、矿物结晶调控和仿生矿化应用三个层面介绍生物矿化的结晶调控原理和取得的重要成就,希望可以作为未来化学工业发展的他山之石。
1 生物矿物-溶液界面结构
1.1 矿物的界面水结构
生物矿物处在水溶液环境,矿物表面与水分子会发生相互作用,形成不同于各自体相结构的界面层结构。矿物表面暴露原子存在不平衡悬挂键,具有较高的反应活性,会与水分子形成氢键或配位键,甚至将水分子劈裂,使表面官能团质子化或羟基化,以降低界面自由能。矿物表面也因此带残留电荷或极化,可改变表面水分子层的偶极取向和氢键网络结构,这种界面效应可以通过水分子氢键网络传递到更远的地方,进而形成多层的水化层结构。当然界面作用是相互的,晶体表面也会发生原子晶格畸变,形成结构弛豫层。本文主要关注水化层结构。
探测水化层结构的主要方法有表面X射线分析技术(X-ray reflectivity, XRR; grazing incidence Xray diffraction, GIXRD)、固态核磁共振(solid-state NMR, ssNMR)、调频或力调制原子力显微镜(frequency/force modulation atomic force microscopy,FM-AFM) 以 及 分 子 模 拟(molecular dynamics simulation, MD; Monte Carlo simulation)。下面列举几种典型的生物矿物水化层结构。
骨和牙的主要无机成分是碳酸化的磷灰石(carbonated apatite, CAP), 羟 基 磷 灰 石(hydroxyapatite, HAP, Ca10(PO4)6(OH)2)和氟磷灰石(fluorapatite,FAP,Ca10(PO4)6F2)常作为其模型晶体被广泛研究。表面X射线分析技术需要大面积平整的晶面,这限制了该技术在HAP 体系的应用,实验中常用FAP作为生物磷灰石的模型晶体。FAP晶体的最大暴露面是FAP(100)晶面,Park 等[5]通过XRR 技术发现,FAP(100)晶面存在两个结构化吸附水层和一个体相层状水层,认为结构化水层的形成与表面磷酸根与水形成的氢键有关。第一结构水层氧原子(OW)与FAP最外层氧原子(OP)间距为2.1Å(1 Å=0.1 nm), 小于水分子的范德华直径(2.82 Å)以及水径向分布的第一配位层(3.2 Å),意味着该水层可能是高度取向构型的结构水层。XRR 技术只能获得垂直于晶面的纵向电子密度分布,平行于晶面的侧向电子密度可以依赖GIXRD 技术进行表征。Pareek 等[6]通过GIXRD 发现,FAP (100)晶面第一吸附水层与钙配位,第二吸附水层与磷酸根形成氢键。
HAP 晶体尺寸通常在亚微米级,难以通过表面X 射线分析技术来探测界面结构,但ssNMR 技术不依赖大尺寸晶体,相反,纳米尺寸的晶体具有大的比表面积,还有利于提高信噪比。ssNMR 可以测量出两个原子核之间的间距。骨矿物(CAP)的ssNMR研究表明[7],1H-31P 间距为2.32~2.55 Å,小于磷灰石晶体中的羟基和磷酸基团的1H-31P 间距,因此这里的H 应该来源于界面水,提供了界面结构水以及结构水与表面磷酸根之间形成氢键的直接证据。具体的界面水结构的三维空间分布需要通过分子模拟来辅助了解。MD 分子模拟显示HAP(001)和(100)的界面结构水层具有偶极取向性(图1)[8],与HAP晶面的表面电荷有关。
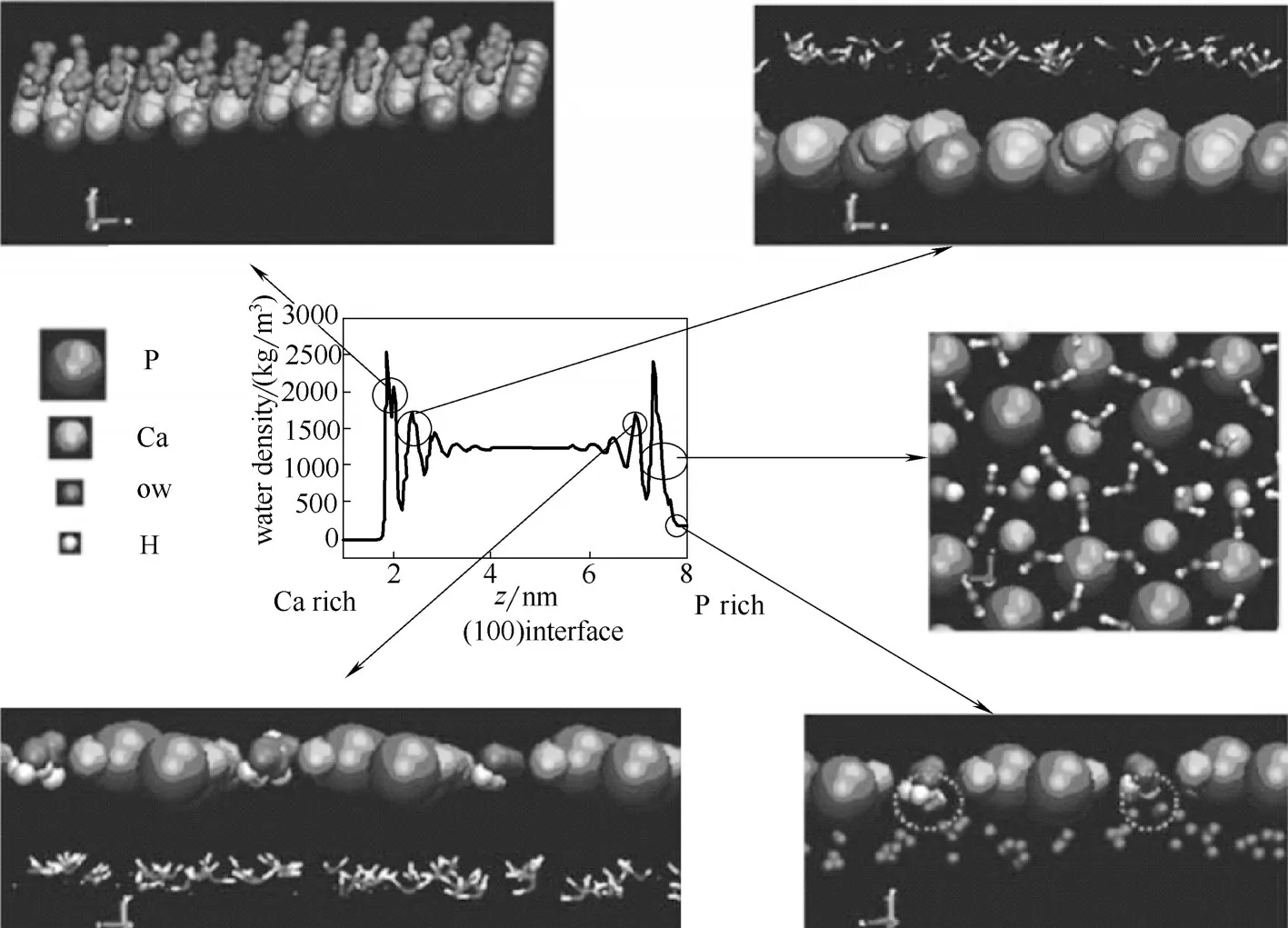
图1 HAP(100)界面水结构[8]Fig.1 Structure of water layers on HAP(100)face [8]
透磷酸钙(brushite,DCPD,CaHPO4·2H2O)是肾结石的主要矿物之一。与HAP 不同,DCPD 晶体带有结晶水。DCPD 结晶水处在钙-磷酸根矿物层之间,具有高度的结构性。在水溶液环境,结晶水暴露在水环境,结构发生重排,形成界面水层,不同于原来的结晶水的结构。Arsic 等[9]通过表面X 射线分析发现DCPD(010)-溶液界面也存在双层结构水,第一结构水层与结晶水类似,第二结构水层在侧向(平行于晶面方向)失去有序性,仅存在纵向的层状分布特点,认为该结构特点可能是DCPD 溶解度低的原因(高溶解度晶体的界面水结构有序度更高[10-11])。DCPD 的双层结构水特征也在MD 分子模拟中重现[12]。MD的分子轨迹分析发现,第一结构水层与外层水的交换律要远小于第二结构水层。
碳酸钙是贝壳中的常见矿物,其中热力学最稳定的物相是方解石晶体(calcite, CaCO3)。XRR 实验发现方解石{104}晶面有两层明显的结构吸附水层,第一结构水层与表面钙结合,距离表面2.3~2.4 Å,第二结构水层与表面碳酸根结合,距离表面3.2~3.5 Å[13]。在XRR实验数据中,第三结构水层(层状体相水)信号不是很明显,但FM-AFM 实验发现三层的结构水[14]与MD 模拟的结果相近[15]。FM-AFM 是目前为止最先进的三维界面结构检测方法,是深入了解水分子和分子离子在晶面的具体吸附位点的重要工具,这部分内容将在1.2节和第2节详细介绍。
以上主要展现的是完美晶面上的界面水结构,但在实际应用体系中,晶体的结晶性可能不好,或者溶液中还存在其他离子或分子,情况更为复杂。在这些复杂体系中,也存在结构水。晶体通过表面弛豫重构,或者溶液中的分子、离子会与水分子形成复合膜吸附在晶体上[16-17]。结构水层和分子吸附层在晶体生长、溶解和聚集中扮演重要的角色,这部分内容将在1.3节作具体介绍。
1.2 界面水结构与水分子吸附位点
矿物界面水层,特别是第一结构水层,往往是晶体与水分子的强烈作用导致的。其中常见的是金属离子与水的配位作用,以及水分子与晶体表面官能团的氢键作用。整个分子水层与晶体表面也可能共同形成氢键网络。这些因素将共同影响界面水的取向结构和具体的吸附位点。
方解石的解理面是{104}晶面,容易获得平整表面,实验研究结果较多。结合当前最先进的表面X射线分析技术[13]、FM-AFM 的实验结果[14]和分子动力学模拟结果[15],目前对方解石{104}晶面的界面水分子吸附位点有了较为统一的认识。图2 是结合不同实验结果和MD分子模拟共同给出的方解石{104}晶面的水分子吸附位点。第一结构水层与表面钙结合。具体位点不在垂直于晶面的正上方,而是侧向偏右。在此位点,水分子的氧正好与表面钙离子形成六配位[图2(a),(b)],与方解石晶体中的钙离子的配位形式一致,该吸附位点也正好占据原来晶体碳酸根的氧位点;第二结构水层位于表面碳酸根上方,与之形成氢键。该位点正好占据原来晶体钙离子的晶格位点。FM-AFM 观测到的第一和第二结构水层的分子图像(探针振动的振幅变化图)与晶体表面的格点对称性特征一致[14]。以上侧向和纵向的结构特征共同说明水分子在方解石{104}晶面的吸附存在固定吸附位点。力场优化过的MD 分子模拟结果几乎可以重现实验中的发现[15][图2(c)]。

图2 方解石{104}界面水吸附位点Fig.2 Adsorption site for water layers on calcite{104}face
关于FAP (100)晶面的水分子吸附位点报道不多,Pareek 等[6]结合GIXRD 和MC 分子模拟结果给出了一个界面水结构模型。研究发现FAP(100)晶面第一吸附水层与钙离子结合,第二吸附水层与磷酸根形成氢键。虽然吸附水分子具有严格的吸附位点,但具体占据原晶格的位点不如方解石体系明确。只能大致看出第一吸附水层占据在原晶格钙离子位点附近,第二吸附水层占据在原晶格磷酸根位点附近。FAP(100)的第一、第二吸附水层与整个晶面形成氢键网络,共同稳定FAP 暴露的晶面,降低了FAP表面的原子畸变。
界面水分子在矿物晶面上有特定的吸附位点:在侧向方向,水分子的吸附位点与晶体表面晶格位点有对应关系,在纵向(垂直于晶面)方向,界面水结构还与晶体结构有一定匹配关系。通过对比HAP(001)和方解石(104)界面水结构与晶体结构(图3,未发表实验结果)发现,界面水结构的高密度区和低密度区,与晶体结构具有一一对应的匹配关系。这说明晶体的结构信息,可以通过晶体表面离子与水分子作用(配位或氢键作用)以及水分子之间的氢键网络传递到晶体-溶液界面。
1.3 界面水结构与吸附能垒
晶体界面的吸附水层是非常牢固的,分子行为接近固态,被认为是“类冰”结构水[8]。探针在接近晶体时突破这些层状结构水需要克服一定能垒,造成探针共振频率或振幅的改变(FM-AFM 探测到水层结构的机理)。这些结构水层的存在将影响分子的吸附、晶体生长中的离子输运,以及纳米晶体之间的聚集行为。

图3 矿物晶体界面水结构与晶体结构的对比Fig.3 Comparison of interfacial water structure with crystal structure
分子突破界面水层吸附到晶面上需要克服多重能垒,所以有的体系通过常规MD 分子模拟在几十乃至几百纳秒的时间尺度也观察不到生物分子或添加剂分子直接吸附在晶面上,往往是隔着一到两个结构水层。能垒的极值点一般处在水分子层间的低密度区,为了使分子克服能垒,可以借助温度加速分子动力学方法、伞状抽样(借助弹簧将分子限定在特定位置)法、拉伸分子动力学方法(steered MD)、metadynamics 方 法、副 本 交 换 法(replica-exchange)等方法[18]。MD 分子模拟结果显示,AFM 探针直接接触晶面需要克服约120 kJ/mol的能垒(根据论文中的力-距离曲线积分计算得到[19]);天门冬氨酸分子进入透磷酸钙{010}晶面需要克服12~25 kJ/mol 的能垒[12]。水分子本身跨越结构水层也需要克服能垒,自由能计算表明,分子进入HAP(001)晶面需要克服约4 kJ/mol的能垒[20]。
在晶体生长中,结构水层将影响溶液中的离子进出晶面,甚至可能成为晶体生长和溶解的速控步骤。Dove 等[21]通过晶体溶解动力学数据分析认为,硫酸盐中阳离子脱离界面水是溶解的速控步骤。该想法得到MD 分子模拟的支持。Piana 等[22]在MD中发现,在长达100 ns的时间尺度,未观察到钡离子突破重晶石(barite,BaSO4)(001)结构水层,但硫酸根可以。伞状抽样自由能计算表明,钙离子突破(001)晶面结构水层的能垒高达20 kJ/mol。
晶界面的结构水还影响晶体的聚集。MD 分子模拟显示,排出方解石{104}晶面间的结构水,需要克服约50 kJ/nm2的能垒(未发表实验结果),可见希望通过压力直接使两个宏观(大于1 mm2)稳定晶面融合几乎是不可能的(但不排除通过离子迁移的方式进行晶界面迁移)。
2 矿物晶面的分子识别
2.1 生物小分子
生物体内存在大量游离的生物小分子,比如氨基酸、柠檬酸、磷脂等,它们会与水竞争,共同吸附在晶面上。生物小分子与人工合成磷酸钙晶体的相互作用研究揭示了其中的吸附机制、识别位点和矿化调控功能。MD 分子模拟研究表明,氨基酸吸附在HAP(010)和(001)晶面,不会破坏界面原来的层状结构特征,只是层状分布的峰值位置有少量偏移[23-24]。这是由于部分界面水被氨基酸取代,氢键网络结构有微小调整引起的。氨基酸中带电荷基团(氨基、羟基和羧基)在晶面上有特定的吸附位点。氨基主要占据原来晶体中的钙格点位置,与HAP中的氢氧根或磷酸根形成氢键;羧基与HAP表面的钙离子作用。这种多点相互作用将大大增强氨基酸与HAP 间的相互作用。从侧向分布来看,氨基酸在HAP 晶面形成特定的吸附斑图,与晶体结构有一定的匹配性。谷氨酸在HAP(001)晶面的吸附自由能约400 kJ/mol,远大于水的吸附自由能(约2.5 kJ/mol)。因此,当HAP 表面位点被谷氨酸占据时,将抑制晶体的生长。MD 分子模拟发现,谷氨酸在HAP(001)晶面吸附自由能要明显大于(100)晶面,因而对(001)晶面具有更强的抑制效果。该研究解释了为什么在谷氨酸的调控下,容易获得片状的HAP,而在空白实验中,HAP 通常是沿c轴生长的针状晶体[23]。这些研究表明,简单的氨基酸分子对HAP 晶面具有识别作用,可以通过各向异性界面吸附调控矿化。
柠檬酸与HAP 有强烈作用,导致柠檬酸在人体主要分布在骨骼中[25]。AFM 实验发现[26],柠檬酸在HAP(100)晶面上具有特定的吸附斑图(图4)。结合MD 分子模拟,最终确定了柠檬酸通过单羧基与单个钙位点结合的作用模式,澄清了之前对柠檬酸和HAP 之间可能的多位点作用模式的猜测。柠檬酸与磷酸钙强烈作用,可以调控不同磷酸钙矿物的界面能,促进磷酸钙DCPD 晶体向HAP 晶体的相转变[27]。
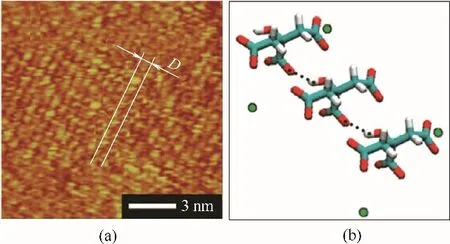
图4 柠檬酸在HAP(100)晶面的吸附位点[26]Fig.4 Adsorption sites of citrates on HAP(100)surface [26]
2.2 蛋白质
结构生物学和生物化学研究表明,矿化功能蛋白质(比如OPN, 骨形态发生蛋白,BMP;牙釉原蛋白,amelogenin;胶原蛋白,collagen等)在生物矿化过程中起着重要的作用[27-30]。这些蛋白与矿物间的相互作用和分子识别引起人们的广泛兴趣。
牙釉质是由针棒状HAP 组成,牙釉原蛋白被认为在其中起着重要的作用[30-34]。ssNMR 实验发现,牙釉原蛋白中的终端羧基与HAP 矿物的距离最近[35]。LRAP 是牙釉原蛋白中包含终端羧基的多肽片段,一旦终端羧基被切除,LRAP 与HAP 的结合就大大削弱[36]。MD 分子模拟结果显示,羧基与HAP表面钙离子的结合是LRAP 吸附在(001)晶面上的关键化学键[37]。
骨形态发生蛋白-2(BMP-2)是诱导成骨重要因子,成骨后仍保留在骨组织。在磷酸钙矿物中添加BMP-2 可诱导骨形成,已临床应用[38]。添加BMP-2的磷酸钙材料可望替代自体骨移植材料,具有很好的应用前景。了解HAP 与BMP-2 的分子作用方式将有利于植入材料的设计和制备,减少BMP-2在体内溶液环境的流失,提高其生物利用度和生物活性。MD 分子模拟研究表明[39],BMP-2 与HAP 存在羧基、羟基、氨基等多位点作用,但极性基团是通过界面水桥连的氢键作用模式,相互作用不如LRAPHAP 强烈。对吸附蛋白质进行拉伸测试中,LRAP分子可严重形变拉长,但羧基仍牢牢抓住HAP 晶面;BMP-2 则在旋转至长轴方向后(蛋白结构并没有明显形变)即与HAP 分离。如何在分子水平改造BMP-2 蛋白,提高结合力还需要进一步深入探讨。
细胞外基质是细胞吸附在生物材料表面的桥梁。细胞分泌的细胞外基质蛋白首先会作用在骨矿物表面,比如纤连蛋白和胶原蛋白。纤连蛋白第三结构域第十模块(FN-III10)富含RGD 残基序列,是与细胞结合的重要位点,被广泛研究。MD 分子模拟研究表明[40],FN-III10 蛋白表面附近的极性和带电荷残基会靠近HAP,疏水残基会远离HAP,引起蛋白结构的部分二级结构改变。在HAP/FNIII10相互作用中,带电荷的羧基和氨基与HAP作用起主导作用,是其他极性基团或电荷基团(如胍基,羟基)作用的两倍。胶原蛋白除了是细胞外基质外,还是骨矿物形成的模板分子。结合原子力显微镜的动态力学谱测量和MD 分子模拟结果,发现胶原与不同磷酸钙晶体之间具有多位点作用和取向识别,但缺钙磷灰石与胶原分子间的取向识别行为可能最接近骨矿物的情形[32]。
以上蛋白与HAP 作用的主要方式是静电力和氢键,但作用强弱有很大差别:有的蛋白可以突破结构水层直接与晶面作用,吸附力较强;有的是通过界面水间接作用在晶面上,吸附力较弱。蛋白质水化层与矿物水化层的相互作用,以及蛋白质如何突破界面水的分子结构关键特征还有待进一步探讨。
2.3 分子组装体
生物矿化中的结构蛋白都富含重复性结构单元,例如骨胶原蛋白富含GXY 序列,是三螺旋蛋白,可组装成纤维结构;贝壳中含有类蛛丝蛋白,富含GA序列,可形成β折叠结晶区域。这些蛋白可能通过组装成有序结构,协同识别晶面。
实验发现,通过构建分子自组装膜(SAM),可以成功调控碳酸钙体系的取向结晶。结晶实验和分子模型/模拟显示,分子组装膜的表面晶格[41]、电荷密度[42]、立体取向[43-44]等因素都可能调控碳酸钙晶体的取向(图5),它们二者在结构特征上具有立体匹配和几何相似性。

图5 SAM膜与方解石{012}面的结构匹配关系的3D模型(不同视角观察)[44]Fig.5 3D models of calcite crystals nucleated with their{012}faces on SAM on Au(111)[44]
Hartgerink 等[45]通过构建双亲多肽自组装纤维来模拟胶原纤维,实现了HAP 晶体的有序矿化。通过自组装,多肽酸性官能团之间形成周期性间距,被认为可能作为HAP成核和外延生长的模板[45-46]。
嵌段共聚高分子可形成胶束,增强与晶体的相互作用。富含羧基的嵌段共聚物(PSPMA30-PDPA47)可在方解石生长过程中埋入晶体中[47]。原位AFM 实验表明,共聚物胶束可选择性吸附在方解石台阶[48]。
一些二维材料具有平整的表面,非常适合原子及亚原子尺度的AFM 观察。在这些体系中发现大量的分子组装体与二维材料界面的有序组装和分子识别,例如云母和胶原[49]、石墨和多肽分子[50]、二硫化钼与多肽[51]等。这些体系分子识别位点和识别机制可能有助于理解生物分子组装体与生物矿物的界面识别。
2.4 矿物界面的手性识别
矿物与分子手性有不解之缘,手性分子的吸附可以导致手性矿物的形成,反过来,手性矿物晶面也可以识别和分离手性分子。在生物世界的手性起源争论中,也有学者认为可能是由某些地质矿物对生物分子对映体的不对称富集引起的。而在生物手性环境中产生的生物矿物往往具有螺旋手性的形态,被认为与生物分子和矿物之间的手性识别密切相关。
研究表明,手性的天门冬氨酸(Asp)可以破坏方解石{104}晶面上台阶的镜像对称性,形成手性的台阶形貌和宏观形貌[52]。MD 分子模拟计算发现,DAsp 手性选择晶面台阶的(014̅)上沿面,L-Asp 手性选择晶面台阶的(11̅4̅)上沿面。这种选择性吸附将在微观上改变晶体台阶生长界面能,从而导致宏观晶体形貌的改变,呈现手性的形貌特征。
手性分子除了通过改变台阶生长影响晶体形貌,还可以通过表面吸附影响晶体的聚集或表面二次成核。手性的谷氨酸(Glu)能导致部分氟取代的HAP(FHAP) 聚集形成束状FHAP, 其中晶体簇的一头呈扇形略微散开,但消旋谷氨酸则在晶体簇的两头都形成扇形[53]。手性的Asp 和Glu 可导致球霰石(碳酸钙的一种晶型,常见于贝壳的珍珠质层)聚集形成复杂的手性螺旋线结构,D 对映体形成顺时针螺旋体,而L 对映体形成逆时针螺旋体[54]。这种螺旋体的产生是由于球霰石片状晶体通过倾转错位堆叠形成的(相邻晶体间有4°的倾角)。分子模拟显示L-Asp 的碳酸根基团更靠近球霰石(100)晶面的钙格点方框的左侧吸附(能量上与右侧吸附位点有0.64 kcal/mol 的差别, 1 kcal = 4.184 kJ),这种不对称吸附可能是造成球霰石片状晶体倾转错位堆叠的分子原因。当然,D-Asp 在晶面的吸附位点选择性是与L-Asp 的吸附位点呈镜面对称的,所以相邻晶体的倾角方向与之相反。
以上生物分子对矿物晶面的手性选择的理解是静态的,它们之间具有一一对应关系。但也有一些独特的手性动态选择现象。研究发现,晶体手性选择性可随着晶体表面台阶的生长和消退呈现动态变化。在过饱和条件,透磷酸钙选择性吸附DAsp,而在欠饱和溶液,透磷酸钙选择性吸附LAsp[12]。这是由于台阶构型的稳态结构在过饱和、欠饱和状态发生变化引起的。MD 分子模拟显示,DAsp倾向于吸附过饱和台阶(step A),而L-Asp倾向于吸附欠饱和台阶(step B)(图6)。
3 矿物结晶调控与仿生矿化
3.1 结晶成核调控
生物矿化可以理解成生物系统通过生物分子进行程序化调控矿物结晶的过程。结晶过程主要包括晶体成核、晶体生长及聚集。其中结晶成核决定了晶体的晶相、成核密度、结晶取向和晶核位置分布,是结晶过程最早也是重要的第一步,但也是目前了解最少的过程(限于观测技术壁垒)。

图6 D-和L-Asp在DCPD台阶([101]step A 和step B)的吸附自由能曲线(a);Asp在台阶上的稳定吸附构型(b)[12]Fig.6 Free energy profiles for adsorption of D-and L-Asp on[101]step A and step B(a);Snapshots of stable configurations in adsorbed state at free energy minimum(b)[12]
目前对结晶成核的理解可以划分为经典成核理论和非经典成核过程[55]。经典成核理论假设成核过程是基于原子聚集的链反应过程,理论基于原子聚集获得的体相自由能减少与新生成物相的表面自由能增加建立准静态平衡关系,得到成核能垒与界面自由能及过饱和度的关系,其中,界面自由能是成核能垒的指数项,对成核起关键调控作用[56-57]。一般认为,生物分子通过调控成核界面能来进行结晶调控。在碳酸钙/SAM 结晶体系中,碳酸钙的结晶成核动力学数据分析结果表明以羧基为端基的SAM 膜可以大大降低成核界面能,加速结晶成核[57],符合经典成核理论的描述。酵母细胞也可通过层层自组装(LbL)技术在表面修饰聚电解质,成核动力学实验表明,细胞修饰后可大大降低成核界面能,促进磷酸钙的成核,使细胞壳化工程成为可能(即在过饱和溶液,矿物优先在细胞表面沉积,而不是在体相溶液)[58]。生物分子在亚稳态晶体表面吸附,还可以改变晶体相变界面能,促进晶体的转化。研究表明,柠檬酸和酸性氨基酸的存在可以加速DCPD向HAP相转化[27]。晶体/溶液的界面能测量结果显示,这些生物小分子可以显著降低相转化的界面能,从而促进相转化动力学,符合经典成核理论的描述。
矿物结晶成核还可以经由其他非经典途径形成,例如经由无定形矿物、液态前体、离子团簇或聚集体或者其他前体物相[55]。生物矿化过程被认为存在多种非经典成核途径[55]。这些途径的存在,打破了基于原子聚集的链反应的经典成核理论假设,因而对这些过程的成核规律和调控机制需要重新认识。研究发现,HAP 经由无定形磷酸钙(amorphous calcium phosphate,ACP)的成核,在高pH(高过饱和度)溶液条件的结晶成核速率反而更慢的反常现象,不符合经典成核理论的描述[59]。成核动力学的系统测量为成功解释该现象奠定了基础[59-60]。研究表明,当ACP 形成后,体系形成ACP 饱和溶液,这时溶液的有效过饱和度不依赖溶液的初始浓度而接近常数,但不同钙、磷酸根的初始浓度决定了ACP的形成总量。矿化过程跟踪发现,HAP 总是优先成核于ACP 表面;再从化学计量关系来看,ACP 转化为HAP 还需要从溶液中摄取额外的钙离子。因此,成核速率与ACP 表面积以及溶液中游离的钙离子相关。在高pH 溶液环境,PO3-4所占比例大大增加,导致活度更高,为保持ACP 的活度积(ACP 相和溶液相之间的化学位平衡),钙离子活度实际上更低,这是导致HAP 成核速率低的重要原因。这种经由无定形物相的矿物成核给结晶调控带来更多的调控通道。例如,可以通过二氧化硅[61]、柠檬酸或高分子包裹ACP 表面抑制HAP 结晶成核[62],可以通过增加ACP 颗粒的分散度或减小ACP 尺寸增加表面积来促进结晶成核[63],还可以通过引入镁离子[64]和锶离子[65]增加HAP 成核需要额外克服的晶格扭曲能来抑制成核等。
3.2 晶体生长和组装调控
晶体的生长是晶体在原有晶体材料表面上继续堆积晶体组分的过程。堆积的具体过程是很丰富的,可以是经典的一个一个分子原子堆积在台阶;也可以是以离子团簇和前体的形式黏附在表面再进行重构[66];还可以是以纳米晶体或纳米颗粒的形式聚集,再进行结构调整[55]。而生物分子和功能分子作用在晶面上或晶体生长基元上,势必会大大影响该堆积过程,从而影响晶体的生长,调控其形貌。
在晶体表面进行分子堆积生长的途径一般认为是分子从体相溶液经界面层到达晶体台阶面(terrace),在表面进行二维扩散到达台阶(step)处(这里的自由能更低),再沿着台阶进行一维扩散,到达台阶扭折(kink)处(自由能最低)[56]。如果异质分子吸附到晶体表面或step/kink 处,将占据晶体组分生长的位点。如果移除异质分子比水(溶剂)分子难,那么晶体生长的速度将被延缓;反之,晶体生长速度还能加速(这里异质分子起催化剂的作用)。如果异质分子的吸附自由能比晶体组分的吸附自由能还低,那么晶体在此处的位点将极可能被异质分子占据(被占据的概率符合热力学Boltzmann 分布,与吸附自由能差呈指数关系),从而形成包晶或晶格掺杂,占据位点附近晶格将形成晶格扭曲。AFM原位观测发现,一种富含电荷的高分子嵌段共聚胶束可以嵌入方解石晶格,并在埋入胶束时,在其上方留下空穴。这些空穴会造成晶格扭曲,可能是提升方解石/高分子复合材料力学性能的重要原因[48]。生物小分子也可以影响晶体的台阶生长。AFM 原位实验发现,加入Asp 后,方解石{104}晶面上的正台阶变得圆钝,许多高能面(针对无添加剂条件而言)暴露,台阶间距也变密,这与Asp 降低台阶能有关,可能是由于Asp 在台阶侧面形成有序吸附层有关[67]。研究发现,Asp 还可加速重晶石的生长[68]。MD 分子模拟的自由能计算表明,Asp可降低钡离子在(001)晶面的脱溶剂能垒——重晶石生长的重要速控步骤。在这过程中,Asp 起到携运阳离子的催化作用[68]。硼酸对石膏(二水硫酸钙)的晶体生长具有双重调节作用,在低浓度下,硼酸促进石膏的晶体生长,但在高浓度抑制其生长。AFM 原位观测发现,随不同浓度添加,硼酸一方面可促进(010)晶面(最大暴露面)的台阶生成速度,另一方面,抑制了台阶前进的速度[69]。二者综合的效果解释了硼酸对晶体生长的双重调节作用。MD 分子模拟显示,硼酸分子可以作用在晶体界面层,从而降低晶体/溶液的界面能,促进表面台阶的成核;但石膏的离子生长位点也同时被硼酸分子占据,因此晶体生长(台阶的前进)也会被抑制。
以分子团簇或纳米前体形式的堆积生长过程,在硅质岩-1 的(010)晶面上被原位AFM 直接观测到[66]。这些前体需要通过结构重构才能与原晶体在晶格上融为一体。这种堆积生长过程也在磁铁矿体系(原位电镜观测)[70]、球霰石体系(离位电镜的过程捕捉)[71]中发现。细致的晶格融合动力学研究表明,晶界迁移可能是结构重构的一个重要途径,而表面吸附形成的界面压力可能是降低晶界迁移能垒的主要驱动力[71]。纳米晶体还可以通过有序聚集的模式进行生长[72-73]。晶体的聚集方式可以通过有机/生物分子进行调控。研究发现,在甘氨酸调控下,纳米磷酸钙可组装成针棒状HAP,在谷氨酸调控下,组装成片状HAP[74]。MD 分子模拟结果显示,甘氨酸分子的吸附升高了HAP(001)晶面的界面能,而谷氨酸则降低了该晶面的界面能,这可能是造成纳米磷酸钙各向异性组装的主要原因。晶体的生长模式可通过分子进行调控。在双亲分子C18-Glu的调控下,方解石的晶体生长模式可以从传统的基于离子堆积的生长模式切换为通过前体聚集的生长模式[75]。研究认为,C18-Glu 可以稳定所形成的碳酸钙前体,并在表面形成保护层,阻止碳酸钙离子通过离子堆积的方式在前体或方解石表面继续生长。
3.3 仿生矿化
生物矿物的典型结构特征是具有多级有序的结构,如何应用分子的结构信息,实现超级有序复合结构的形成是仿生矿化研究的关键科学问题。在生物矿化仿生结构制备中,类牙、类骨和类贝壳是研究较多的体系。
牙釉质材料的结构特征是成捆的针状HAP 晶体有序聚集在一起。在天然牙釉矿化过程中,牙釉原蛋白被认为在其中起重要的结构诱导作用。在体外模拟实验也发现,牙釉原蛋白可帮助纳米磷酸钙进行有序组装[74,76]。超长HAP 纳米线可以通过油酸的表面修饰在乙醇溶剂中经疏水作用有序聚集在一起,形成类牙釉质结构[77]。如何在天然牙釉质表面组装上类牙釉质的结构材料对于牙修复而言是一个重要的挑战。实验发现,牙釉原蛋白有助于形成类牙釉修复层[78-79];在高浓度谷氨酸(100 mmol/L)的调控下,20 nm HAP 可以在牙釉质表面有序组装成类牙釉质结构。谷氨酸可能作用在HAP(001)晶面,在HAP 组装中起桥连的作用[80]。牙釉质材料中的HAP 晶体还可以在原来晶面的基础上继续外延生长。通过模拟生物矿化中的矿化结晶前沿结构,在牙釉质晶体表面形成致密的ACP涂层,可以在模拟口腔液态环境进行外延生长,形成2~3 μm的修复层,新生成的修复层结构几乎与天然牙釉质一致[81]。
贝壳材料具有超高的强度和韧性,也是生物矿化仿生学习的热点。贝壳结构分为内外两层,外层为柱状层结构(晶体呈棱柱状堆积),内层为珍珠质层结构(片状碳酸钙晶体与有机基质形成“砖-泥”结构;片状碳酸钙晶体之间还可能形成矿物桥,进一步增强材料的力学性能)。生物矿化研究认为,棱柱状层结构可能是通过竞争生长形成的。其中,关键性问题是如何在二维基质表面大量形成随机取向的晶种。通过模拟贝壳中的有机基质/碳酸钙复合膜,经过三个合成步骤可实现高度取向的棱柱型碳酸钙薄膜的仿生合成[85]。首先涂覆聚合物基底,形成二维球晶;然后在薄膜上沉积矿物过渡层,仿生形成矿物/基质复合膜;最后是晶体的过生长,通过竞争生长形成有序结构。仿生矿化后的碳酸钙薄膜具有与贝壳相似的结构和相当的硬度及杨氏模量。值得一提的是,聚合物二维球晶可诱导沉积碳酸钙(球霰石)二维球晶,体现出分子结构信息向矿物结构的转化。珍珠质层是贝壳优秀力学性能的主要原因,可以通过仿生矿化得到类珍珠质层结构[86]。首先通过冷冻浇注形成层状的有机基质(壳聚糖),然后将壳聚糖乙酰化,形成甲壳质,再通过矿化液在层间沉积聚合物稳定的碳酸钙矿化前体,然后再渗入丝素蛋白,在热压条件使碳酸钙相变为文石晶体。这里形成的文石层不是单一取向,不同于天然珍珠质层,可能与有机模板不具备长程有序结构有关。
4 结 论
综上所述,矿物晶体表面会形成结构化的水层和分子吸附层结构,其结构与晶体结构有很大的相关性。这种相关性是通过晶体表面金属离子和阴离子通过配位作用和氢键作用传导到第一吸附水层,然后通过第一吸附水层传递到更远的界面层。吸附水层的侧向结构和界面层的纵向结构都在一定程度上传递着晶体的结构信息。反过来,有序组装的生物分子层也会将分子的结构信息和分子立体取向通过界面水传递到矿物离子,这样离子在生物分子模板界面形成特定吸附位点,在结构上可能近似晶体的某个晶面结构,从而导致矿物在分子模板上的取向性成核。
矿物晶面与生物分子的识别作用,可以应用于分子富集、手性分离和生物分子组装调控等,对于生物传感器件、能源转化材料、环境污染治理、分离和催化、超结构复合功能材料的制备等方面具有重要的意义。
生物矿化研究中认识的生物分子对无机矿物晶体的成核、生长和聚集的调控原理,以及通过仿生矿化获得的多级有序材料制备方法,综合体现了生物分子与矿物晶面之间的识别作用和相互调控。通过界面结构信息传递实现分子信息向晶体结构转化是一种高级的分子工程和结晶工程策略,可望应用于未来化学工程以实现复杂结构工程材料的过程调控和大规模制备。
符 号 说 明
ACP——无定形磷酸钙
AFM——原子力显微镜
amelogenin——牙釉原蛋白
Asp——天门冬氨酸
上述农谚即是农民长期经验的总结,不管是农作物栽种、施肥还是五畜饲养都要具体问题具体分析,根据各自的特点,尊重客观规律,做到一切从实际出发。
barite——重晶石
BMP——骨形态发生蛋白
BMP-2——骨形态发生蛋白-2
C18-Glu——十八烷基谷氨酸
CAP——碳酸化的磷灰石
calcite——方解石
collagen——胶原蛋白
DCPD——透磷酸钙
FAP——氟磷灰石
FM-AFM——调频或力调制原子力显微镜
FN-III10——纤连蛋白第三结构域第十模块
GA——氨基酸序列,G为甘氨酸,A为丙氨酸
GIXRD——掠角入射X射线衍射
Glu——谷氨酸
GXY——氨基酸序列,G 为甘氨酸,X、Y 为任意其他氨基酸
HAP——羟基磷灰石
kink——台阶扭折
LRAP——牙釉原蛋白中包含终端羧基的多肽片段
MC——Monte Carlo分子模拟
MD——分子动力学模拟
OPN——骨桥蛋白
PILP——聚电解质稳定液态前体
PSPMA30-PDPA47——一种富含羧基的嵌段共聚物
RGD——氨基酸残基序列Arg-Gly-Asp,可与细胞整合素结合
replica-exchange——副本交换法
SAM——分子自组装膜
ssNMR——固体核磁共振
steered MD——拉伸分子动力学方法
step——台阶
terrace——台阶面
XRR——表面X射线反射
