鳗鲡肠道微生物抗性基因组成特征的初步分析
2020-03-08黄薇刘兰英罗土炎刘洋宋永康
黄薇 刘兰英 罗土炎 刘洋 宋永康

摘要:肠道微生物是抗生素抗性基因(ARGs)的储存库,为明确ARGs在鱼肠道微生物中的组成特征,本研究以养殖鳗鲡为研究对象,利用高通量测序技术及功能宏基因组学分析方法对鳗鲡肠道微生物ARGs的种类和丰度进行了探究。研究结果显示,养殖鳗鲡肠道微生物主要由厚壁菌门(Firmicutes)、梭杆菌门(Fusobacteria)、变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes)组成,比对抗性基因数据库共注释得到254种ARGs基因,分别归属于24类抗性类型,其中高丰度的抗性类型为多肽类抗生素、多重耐药、氟喹诺酮类、四环素类和β-内酰胺类。以上结果表明,在鳗鲡养殖过程中,出现耐多肽类抗生素、氟喹诺酮类、四环素类、β-内酰胺类以及多重耐药病原菌的机会相对较高。本研究为抗生素在鳗鲡养殖过程中合理使用提供参考依据,同时也显示宏基因组测序技术在检测鱼肠道ARGs的可行性和优越性。
关键词:鳗鲡;肠道微生物;抗生素抗性基因;宏基因组学
中图分类号:S182 文献标志码:A 文章编号:1002-1302(2020)21-0057-05
中国是水产养殖大国,水产养殖产量占世界养殖总产量的70%左右,位居世界第一[1]。近年来,随着集约化水产养殖的迅速发展,水产养殖中的病害问题特别是细菌性病害在密集型的养殖体系中发病率极高,导致抗生素被大量过度使用。虽然抗生素具有杀菌、促进生长等作用,但长期重复使用或过度使用不仅会降低抗生素的药效,还有可能诱导产生一系列携带抗生素抗性基因(antibiotics resistance genes,ARGs)的耐药性菌株[2]。ARGs作为一项新型环境污染物在2006年被明确提出[3],成为一个全球性的环境热点问题,由此而产生的潜在生态风险也日益引起各国政府和研究者的广泛关注。
水产养殖鱼类肠道和粪便内有耐药菌和ARGs检出的报道已屡见不鲜[4]。动物肠道本身就是细菌生长繁殖的重要场所,肠道内共生着大量的已知和未知的微生物,环境中抗生素的选择压力容易诱导其内在或外源ARGs通过质粒接合转移等方式传递给肠道内的大量敏感菌群变成新的耐药菌,因此鱼类肠道是ARGs定植和转移的一个非常理想的微环境[5]。研究人员认为,鱼肠道为ARGs的转移和扩散提供了安全而稳定的场所可能是ARGs能够在水环境中长期稳定存在的重要原因[6]。因此,明确养殖鱼体肠道菌群中ARGs的污染特征,对水产养殖环境ARGs的污染评价及生态安全管理具有重要的作用。
传统研究环境ARGs的方法主要是通过耐药菌的培养,以及ARGs的PCR和定量PCR筛选等,通过这些方法研究人员已经从不同环境介质中分离和鉴定了大量的ARGs,阐释了不同ARGs的作用机制[7]。但传统的研究分析方法具有一定的限制性,如大多的微生物不可培养、PCR结果的真实程度取决于设计引物的序列等,因此很难得到环境中微生物抗生素抗性基因组的全面和详细的信息。近年来,基于功能的宏基因组筛选和基于高通量测序为基础的宏基因组测序分析技术与方法的发展为研究环境抗生素抗性基因组的生态、起源、进化和传播机制提供了强有力的工具[8-9]。Looft等[10]和Xiong等[11]对猪和肉鸡肠道微生物组的高通量宏基因组测序分析,使我们对猪和鸡的肠道微生物组所蕴藏ARGs的广度和深度都有了更进一步的理解,同时为微环境下ARGs的传播和流动提供了有力的证据。然而与陆生脊椎动物相比,水生动物所处生态环境更为复杂,其肠道微生物具有更丰富的多样性和复杂性,受环境条件和随机因子的影响,使得鱼类肠道微生物比陆地动物更具动态性,直至目前鱼类肠道微生物ARGs组的相关性研究也鲜见报道。
鳗鲡(Anguilla sp.)是中国出口创汇最多的水产养殖品种之一,年产值超百億元,是目前单项农产品中创汇最多的品种之一。鳗鲡在人工养殖过程中疾病时有发生,其中细菌性疾病的危害最为严重,常造成重大的经济损失。明确ARGs在鳗鲡肠道微生物中的组成特征,可以为指导鳗鲡养殖抗菌药物的合理使用提供必要的理论依据,对水产动物源病害防控具有重要的应用价值。本项目拟以养殖鳗鲡为研究对象,运用高通量测序技术及功能宏基因组学分析方法,确定鳗鲡肠道微生物中ARGs的种类和丰度,为鳗鲡的健康养殖、病害防控以及水生态环境调控参考依据。
1 材料与方法
1.1 样品采集
2019年3月12日,选取福清鳗鲡养殖区的3个典型养鳗场作为采样点,每个养鳗场随机网捕3条鳗鲡,体质量200~300 g,送回实验室。将采集的鳗鲡头尾固定,表面采用75%乙醇消毒90 s,使用无菌去离子水冲洗3次,将每条鳗鲡用无菌刀解剖,取其肠道,立即放入含15 mL PBS缓冲液、1 mL茶树油和20 g石榴石(0.7 mm)的无菌50 mL离心管中,用一次性无菌研杵压碎肠道,在涡流振荡器中以1 500 r/min的速度振荡5 min使其均匀化。将匀浆混合物依次通过100、20、11和8 μm的滤膜过滤,去除宿主细胞,再经过0.22 μm滤膜富集微生物,收集的肠道微生物滤膜储存在-80 ℃下,以提取其总基因组DNA。
1.2 主要试剂
75%乙醇、PBS缓冲液、无菌研杵,购自生工生物工程(上海)股份有限公司;茶树油、石榴石,购自德国Qiagen公司;100、20、11、8和0.22 μm的滤膜,购自美国Millipore公司;PowerWater DNA Isolation Kit试剂盒,购自美国MOBIO公司。
1.3 DNA提取与纯化
取收集好的鳗鲡肠道微生物滤膜,采用PowerWater DNA Isolation Kit 试剂盒提取总基因组DNA,具体提取方法参考试剂盒说明书。提取的基因组DNA样品用Qubit 2.0核酸蛋白定量仪进行定量,然后放置在-80 ℃下保存直至使用。
1.4 Illumina测序及高通量数据处理
为达到测序要求的DNA阈含量,将9个DNA样品混合,由上海美吉生物公司采用Illumina Hiseq 2500测序平台进行宏基因组测序。高通量测序原始数据先采用fastp软件进行质控去除接头序列、低质量碱基、N碱基及长度过短序列,利用Megahit与Newbler软件对质控数据进行多重混合拼接组装,使用MetaGene软件对拼接结果中的Contigs进行ORF预测,采用CD-HIT软件进行聚类(默认参数为:95% identity、90% coverage),每个类取最长的基因作为代表序列,构建非冗余基因集,使用BLASTP软件将非冗余基因集与NR数据库进行比对(比对参数设置期望值e-value为1e-5),并通过NR库对应的分类学信息数据库获得物种注释结果,然后使用物种对应的基因丰度总和计算该物种的丰度,从而构建相应分类学水平上的丰度表,将数据上传ARDB数据库(http://ardb.cbcb.umd.edu/)进行比对(比对参数设置期望值e-value为1e-5),获得基因对应的抗生素抗性功能注释信息;将数据上传CARD数据库(http://arpcard.mcmaster.ca/)进行比对(比对参数设置期望值e-value为1e-5),获得基因对应的抗生素抗性功能注释信息,综合CARD和ARDB的注释结果,绘制鳗鲡肠道微生物基因组抗性基因图谱。
2 结果与分析
2.1 高通量测序序列数据分析
鳗鲡肠道微生物基因组DNA样品经宏基因组测序共得到原始序列(Raw reads)50 839 026条,质控后的有效序列(cean reads)50 355 024条,占原始序列的99.45%。混合拼接后得到Contigs 65 048条序列,N50为404 bp;经过基因预测得到ORFs的序列条数为406 331,通过聚类构建了200 260个基因的非冗余基因集。
2.2 鳗鲡肠道菌群结构和多样性分析
利用BLASTP将非冗余基因集与NR数据库比对进行物种分类注释,共得到71个门水平、147个纲水平、373个目水平、686个科水平和1 473个属水平和4 094个种水平的物种,其中注释为真细菌、病毒、古细菌、真核生物以及未分类物种的序列数分别占总注释序列数的96.13%、0.17%、0.04%、3.64%和0.02%。从注释结果中可知真核生物的序列数比例仅为3.64%,说明本研究提取的鳗鲡肠道微生物基因组DNA样品基本清除了宿主DNA。
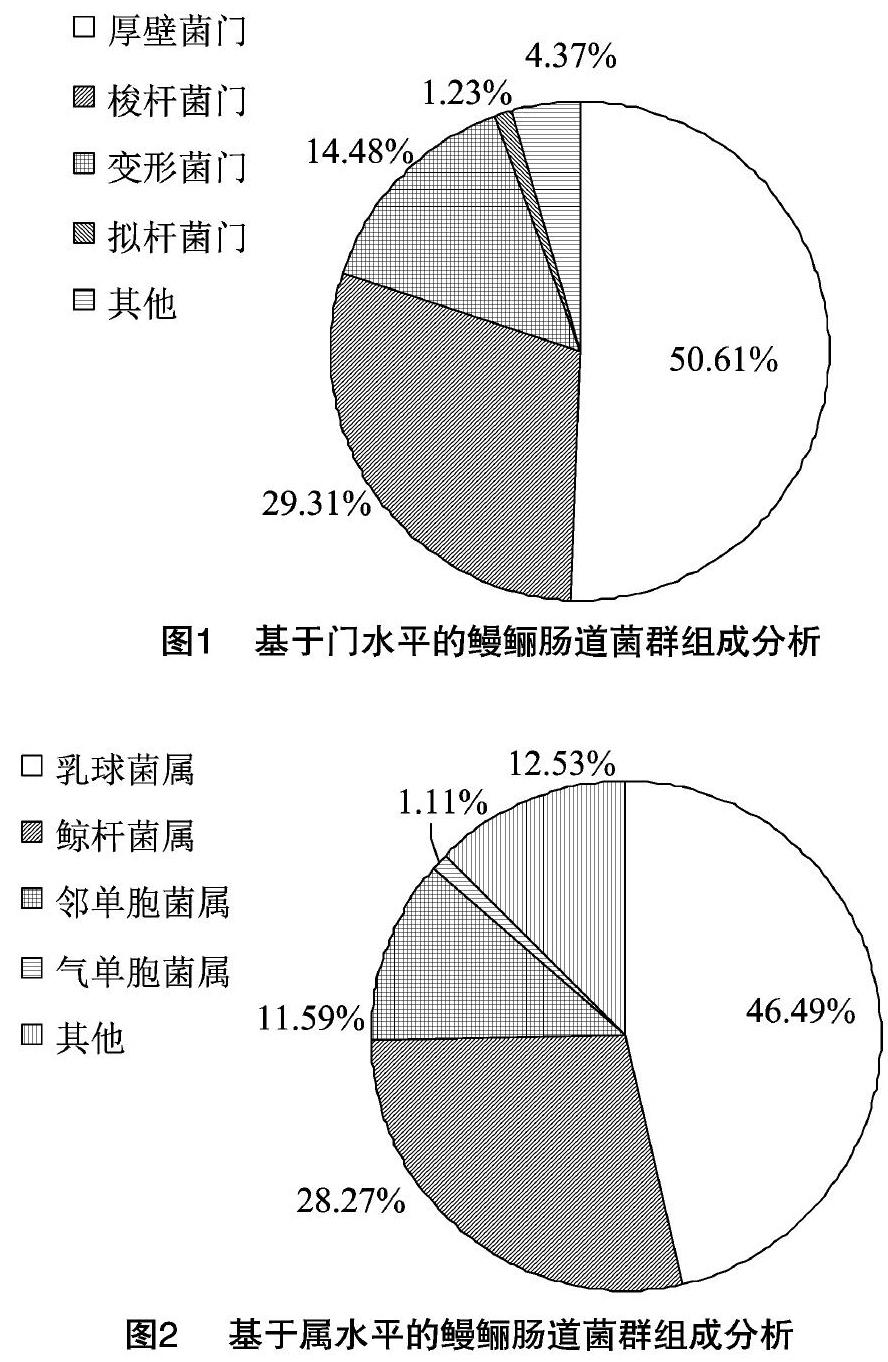
从门水平上看,鳗鲡肠道微生物的优势菌群主要由厚壁菌门(Firmicutes)、梭杆菌门(Fusobacteria)、变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes)组成,其丰度分别占物种注释序列总数的50.61%、29.31%、14.48%和1.23%,除此之外的其他门类的细菌在鳗鲡肠道样品中的丰度均较低(图1)。从属水平上看,鳗鲡肠道微生物中相对丰度在1%以上的细菌属主要有乳球菌属Lactococcus(46.49%)、鲸杆菌属Cetobacterium(28.27%)、邻单胞菌属Plesiomonas(11.59%)、氣单胞菌属Aeromonas(1.11%)(图2)。上述结果使我们对鳗鲡肠道菌群多样性以及优势菌群结构组成有了更全面的认识。
2.3 鳗鲡肠道微生物基因组抗性基因分析结果
将鳗鲡肠道微生物基因组数据上传CARD数据库进行ARGs的预测和注释。结果显示,鳗鲡肠道微生物基因组在CARD抗性基因数据库中共预测得到669 852条ARGs序列,分别属于235个不同的ARGs。不同抗性数据库的侧重点不同,所收录的ARGs序列数目、标准以及注释结果的筛选标准也不相同,导致不同数据比对和注释结果也不尽相同[12]。基因组数据上传ARDB抗性基因数据库,共注释得到27 736条ARGs序列,得到40种ARGs。本研究综合CARD和ARDB的注释结果,对鳗鲡肠道微生物的抗性基因类型进行了统计分析,结果见表1。
由表1可知,鳗鲡肠道微生物中共存在ARGs基因254种,归属于24类抗性类型。具体包括61种多重耐药基因、46种多肽类抗生素ARGs、31种氟喹诺酮类ARGs、23种四环素类ARGs、25种β-内酰胺类ARGs、6种大环内酯类ARGs、9种林可酰胺类ARGs、4种利福平类ARGs、1种Microcin J25 ARGs、5种磷霉素类ARGs、5种Elfamycin ARGs、3种异烟肼类ARGs、4种链阳霉素A ARGs、1种螺旋霉素类ARGs、8种氯霉素类ARGs、9种氨基糖苷类抗生素ARGs、3种磺胺类ARGs、1种羰基氰化物间氯苯腙(CCCP)ARGs、1种夫西地酸类抗生素ARGs、1种阿霉素ARGs、4种甲氧苄啶类ARGs、1种硝基呋喃类ARGs、1种莫匹罗星类ARGs和1种春雷霉素类ARGs。统计结果表明,鳗鲡肠道微生物中存在种类多样且丰富的ARGs库。
在CARD的注释结果中,丰度最高的ARG为氟喹诺酮类抗性基因mfd,丰度最高的抗性类型为多肽类抗生素;在ARDB注释结果中,丰度最高的ARG为四环素类抗性基因tetS,丰度最高的抗性类型为四环素类。综合CARD和ARDB的注释结果,鳗鲡肠道微生物ARGs中较高丰度的抗性类型为多肽类抗生素、多重耐药、氟喹诺酮类、四环素类和β-内酰胺类。表明在鳗鲡养殖过程中,出现耐多肽类抗生素、氟喹诺酮类、四环素类、β-内酰胺类以及多重耐药病原菌的机会相对较高。
3 讨论
环境ARGs的种类和丰度与细菌群落结构组成密切相关[13]。研究表明,不同菌种携带ARGs的偏好性不同,优势菌群对环境ARGs的组成特征扮演着重要角色[14]。宏基因组测序方法不仅可以检测ARGs的广谱特征,同时还能检测出环境微生物群落结构组成。测序分析结果显示,鳗鲡肠道微生物中的优势菌群由厚壁菌门(Firmicutes)、梭杆菌门(Fusobacteria)、变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes)组成,这个结果与之前笔者所在课题组采用16S rRNA基因高通量测序分析得到的优势菌群[15]是一致的,说明本研究的宏基因组测序结果可靠性较高。但2种研究方法得到的优势菌群的分布比例有较大差异,分析原因可能是不同的鳗鲡肠道样本的微生物菌群丰度本身就存在较大差异,同时也可能与不同基因组16S rRNA基因的PCR扩增的特异性有关。此外,鳗鲡肠道的优势菌门和优势菌属与已知斑马鱼[16]、草鱼[17]和虹鳟鱼[18]的肠道优势菌群结果相似,说明鱼类肠道微生物的种类结构相似。
從ARGs注释结果可知,本研究共在鳗鲡肠道中检测出254种ARGs,发现了许多在鱼类肠道未曾报道过的ARGs,如tlrC、YojI、emrB等。导致这些基因未曾报道的原因可能是由于这些基因的宿主(耐药菌)难以培养或是用于扩增这些基因的引物难以设计,这也进一步说明宏基因组测序分析方法比传统方法更能全面的检测出样品中的ARGs[19]。Tamminen等在6年未使用过抗生素的水产养殖场中仍检测到3种编码四环素的ARGs[20]。本研究在鳗鲡肠道中,同样检测出了氯霉素类、硝基呋喃类、磺胺类、β-内酰胺类以及人用抗生素等多种禁用渔药的ARGs,由此可以说明水产养殖场已经成为环境中ARGs的储存库。某些ARGs一旦转移到病原菌中,将使鱼病治疗面临更多的新挑战,同时肠道中的细菌密度极高又极大地增加了基因横向转移的风险,这些ARGs可能通过多种途径(如食物链)最终传递到人体中。
鳗鲡肠道微生物中丰度最高的抗性类型为多肽类抗生素、多重耐药、氟喹诺酮类、四环素类和β-内酰胺类。多肽类抗生素、氟喹诺酮类、四环素类和β-内酰胺类均为水产养殖中常用的动保产品,尤其是多肽类抗生素,由于其添加在饲料中不影响适口性,对水产动物具有促生长、提高饲料转化率的效果,常常作为饲料添加剂广泛使用,大多数的种类都没有残留限量要求[21]。在鳗鲡肠道中,检测出多重耐药基因61种,在CARD数据库比对结果中,这些多重耐药基因占ARGs基因总数的12.95%,基因的多重耐药性已经在许多研究中被证实[19]。抗生素的使用及残留的环境选择压力与抗生素抗性的产生密切相关[3]。鳗鲡肠道中检测出丰度较高种类繁多的多重耐药基因可能与鳗鲡环境中使用的多种抗生素造成的微生物选择压力或者细菌之间ARGs的交换有关[22]。由此可知,水产养殖中抗生素的滥用和过度使用状况不容乐观。本研究采用宏基因组测序分析技术明确了鳗鲡肠道中ARGs的污染特征,可以为指导鳗鲡养殖抗菌药物的监管提供必要的参考依据,对水产动物源病害防控具有重要的应用价值,同时也表明宏基因组测序技术检测ARGs具有的可行性和优越性。
参考文献:
[1]张骞月,赵婉婉,吴 伟. 水产养殖环境中抗生素抗性基因污染及其研究进展[J]. 中国农业科技导报,2015,17(6):125-134.
[2]李云莉,高权新,张晨捷,等. 养殖水域抗生素抗性基因污染的研究概况与展望[J]. 海洋渔业,2017,39(3):351-360.
[3]Pruden A,Pei R T,Storteboom H,et al. Antibiotic resistance genes as emerging contaminants:studies in northern Colorado[J]. Environmental Science & Technology,2006,40(23):7445-7450.
[4]Budiati T,Rusul G,Wan-Abdullah N W,et al. Prevalence,antibiotic resistance and plasmid profiling of Salmonella in catfish (Clarias gariepinus) and tilapia (Tilapia mossambica) obtained from wet markets and ponds in Malaysia[J]. Aquaculture,2013,372/373/374/375:127-132.
[5]Van Reenen C A,Dicks L M. Horizontal gene transfer amongst probiotic lactic acid bacteria and other intestinal microbiota:what are the possibilities? a review[J]. Archives of Microbiology,2011,193(3):157-168.
[6]付佳伦. 细菌耐药基因在斑马鱼体内定植、转移规律及机制研究[D]. 北京:中国人民解放军军事医学科学院,2017.
[7]田宝玉,马荣琴. 环境微生物的抗生素抗性和抗性组[J]. 中国生物工程杂志,2015,35(10):108-114.
[8]Gupta S K,Padmanabhan B R,Diene S M,et al. ARG-ANNOT,a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes[J]. Antimicrobial Agents and Chemotherapy,2014,58(1):212-220.
[9]Gibson M K,Forsberg K J,Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology[J]. The ISME Journal,2015,9(1):207-216.
[10]Looft T T,Allen H K,Bayles D O,et al. In-feed antibiotic effects on the swine intestinal microbiome[J]. Proceedings of the National Academy of Sciences of the United States,2012,109(5):1691-1696.
[11]Xiong W,Wang Y L,Sun Y X,et al. Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes[J]. Microbiome,2018,6(1):34.
[12]田容川,黄 薇. 鸡肠道大肠杆菌多抗菌株C20耐药性基因的预测和分析[J]. 福建农业学报,2017,32(9):932-938.
[13]Guo J H,Li J,Chen H,et al. Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and Mobile genetic elements[J]. Water Research,2017,123:468-478.
[14]Wang J H,Lu J,Zhang Y X,et al. Metagenomic analysis of antibiotic resistance genes in coastal industrial mariculture systems [J]. Bioresource Technology,2018,253:235-243.
[15]Huang W,Cheng Z,Lei Shao Nan,et al. Community composition,diversity,and metabolism of intestinal microbiota in cultivated European eel (Anguilla Anguilla)[J]. Applied Microbiology and Biotechnology,2018,102(9):4143-4157.
[16]Roeselers G,Mittge E K,Stephens W Z,et al. Evidence for a core gut microbiota in the zebrafish[J]. The ISME Journal,2011,5(10):1595-1608.
[17]Wu S,Wang Guitang,Angert E R,et al. Composition,diversity,and origin of the bacterial community in grass carp intestine[J]. PLoS One,2012,7(2):e30440.
[18]Wong S,Waldrop T,Summerfelt S,et al. Aquacultured rainbow trout (Oncorhynchus mykiss) possess a large core intestinal microbiota that is resistant to variation in diet and rearing density[J]. Applied and Environmental Microbiology,2013,79(16):4974-4984.
[19]趙 帝,徐在言,吴山功,等. 草鱼肠道微生物抗生素抗性基因研究[J]. 水生态学杂志,2019,40(6):111-116.
[20]Tamminen M,Karkman A,Lhmus A,et al. Tetracycline resistance genes persist at aquaculture farms in the absence of selection pressure[J]. Environmental Science & Technology,2011,45(2):386-391.
[21]陈奕彬,胡 娟,杨宪宽,等. 抗菌肽在水产养殖中的应用研究进展[J]. 饲料工业,2015,36(12):36-39.
[22]鲁 曦. 低剂量抗生素刺激条件下耐药基因水平传播的机制研究[D]. 广州:华南理工大学,2012:3-5.邓宽平,杨秀伟,杨胜伟,等. 基于SSR标记的树莓资源遗传多样性分析[J]. 江苏农业科学,2020,48(21):62-67.
