染色质构象与基因功能
2020-03-04黄其通李清张玉波
黄其通,李清,张玉波
综述
染色质构象与基因功能
黄其通,李清,张玉波
中国农业科学院农业基因组研究所,“岭南现代农业”广东省实验室,深圳 518120
在真核细胞中,DNA序列以染色质为载体,高度凝缩并存储于细胞核内,其复制、修复和转录表达等过程受到染色质构象的精准调控。越来越多的研究表明,特定的染色质构象可选择性激活或沉默基因,从而控制细胞自我维持或定向分化,决定细胞的组织特异性和细胞命运。因此,对染色质构象的深入研究已成为准确解析基因功能的一个关键切入点,也是当前基因组学研究所面临的一个巨大挑战。本文对染色质构象的研究历史、结构特征、动态调控机制进行了综述,并重点论述了不同维度构象特征对基因转录调控的影响,对该领域的研究难点进行了讨论,展望了其未来的发展方向,期望通过有效梳理染色质构象与基因调控之间的脉络关系,为未来该领域的研究提供参考。
染色质构象;基因功能;动态调控
DNA是生物体的主要遗传物质。在真核细胞中,DNA序列与组蛋白结合形成核小体,并逐渐凝缩为染色质存储于细胞核内[1]。作为细胞核内DNA的载体,染色质的空间构象决定了基因组凝缩的空间特征,从而决定基因组生物学功能的分子基础——调控元件对特定转录因子的招募,及远端调控元件与靶基因的互作[2,3]。与蛋白质结构相似,按不同的空间尺寸,染色质构象可分为3个不同维度(图1):一维结构为核小体的物理定位,反映了不同基因组部位的可接近性;二维结构为核小体串进一步折叠或凝缩形成的染色质构象,如30 nm染色质纤维[4],重点研究邻近核小体之间的相互作用;三维结构为染色质的三维空间构象,是基因组范围内的、广泛的远距离互作结构,包括可能存在的跨越几kb到几Mb距离不等的染色质疆域(chromosome territory, CT)[5]、染色质区室(chromatin compartment A/B)[6],拓扑相关结构域(topologically associating domain, TAD)[7,8],染色质环(chromatin loop)[9],以及反式互作等。近年来,由于高通量测序技术的迅猛发展和多学科的交叉融合,探究染色质构象的新方法层出不穷[10,11],对染色质构象尤其是一维和三维构象的研究不断走向深入,为探索染色质构象背后的生物学意义打下了重要的基础。
现有研究表明,染色质构象具有高度的异质性,不同的细胞状态或表型与特定的染色质构象之间存在着较强的关联性[12]。例如,胚胎干细胞的分化往往伴随着染色体内部互作的逐渐增强[13];癌症细胞中常见compartment A/B转置、TAD边界改变等现象[14,15]。染色质构象开始被科学家们认为是决定遗传信息传递的空间密码[16~18]。生物体通过染色质构象的改变,调控特定基因的复制、修复、转录表达等过程,进而决定细胞功能及其命运[19]。因此,准确捕捉染色质构象,并对其生物学意义进行深入解读,将成为人类进一步认识生命复杂行为的重要基础。
本文将以染色质构象与基因功能为主线,从一维到三维、多角度综述染色质构象的结构和功能特征,并对其可能的动态调控机制进行概述,以期为后续染色质构象与基因功能研究提供参考。
1 染色质的一维构象
核小体是染色质的基本组成单元,最早于1974年由Komberg[1]通过MNase酶切和电镜实验发现。在电镜视野下,核小体与核小体相连,呈现出经典的“串珠”结构(beads-on-a-string)[20,21]。但直到1997年,人们才成功获得核小体的高分辨(2.8 Å)晶体结构[22]。结构分析显示,核小体是由147 bp长的DNA缠绕组蛋白八聚体(组蛋白H2A、H2B、H3和H4各2个分子形成一个组蛋白八聚体) 1.65圈后形成,DNA通过14个小沟与组蛋白八聚体作用形成紧密的结构。

图1 不同维度的染色质构象
核小体的发现与结构的确定是染色质构象研究中的一个里程碑事件,同时也成功开启了从染色质结构到功能的遗传信息解读序幕。相关研究表明,核小体在基因组上的分布有一定的偏好性:核小体在调控区域,如启动子、终止子和转录因子结合位点(TFBS)区域分布稀疏,在编码区分布密集[23~25]。而Saragosti等[26]关于SV40微小染色体的无核小体区和DNase超敏感区定位实验显示,核小体的缺失区域与活跃基因调控区——DNase超敏感区高度吻合。这些结果表明,核小体的定位影响基因表达,具有重要的生物学意义。同时,大量研究也表明了核小体的定位与DNA复制、修复、转录和可变剪接等过程密切相关[27~29]。例如,通过光镊实验观察单个RNA II型聚合酶(RNA polymerase-Ⅱ,RNA Pol Ⅱ)在转录延伸阶段的行为,Hodges等[27]发现编码区的核小体会使RNA Pol Ⅱ多次停顿,导致转录速率下降。而酵母启动子区域核小体的分析结果显示,启动子区域核小体的数量与启动子下游基因转录速率呈负相关关系[25]。
纵观目前研究进展,核小体定位影响基因表达的主要作用模式可概括为两类:一是识别标记,二是接触阻碍。通过研究核小体定位与转录起始位点(TSS)的关系,科学家们发现,启动子区域存在稳定开放的核小体缺乏区域(nucleosome free region, NFR),RNA Pol Ⅱ停靠在NFR的下游+1核小体处,起到调控基因转录表达的作用[30,31]。对于缺乏核心启动子元件的基因,转录复合物依然可正确识别转录起始位点,但在不改变核心启动子序列的情况下,替换NFR,却会导致TSS位置发生偏移[32,33]。这暗示着核小体定位很可能是转录机制准确识别TSS的一个重要标记。此外,也有相关证据表明,核小体定位与可变剪接过程中外显子的识别有关。如外显子上的核小体丰度明显高于内含子、假外显子区域,且丰度越高,剪接信号越弱[34,35]。但总体而言,目前对于核小体识别标记功能的具体机制尚不明确,有待进一步研究。核小体的阻碍效应主要与核小体的自身元件组蛋白密切相关。重构染色质的体外转录实验显示,当裸露的DNA与核心组蛋白(H2A、H2B和H4)形成核小体,并达到每200 bp DNA一个核小体的密度时,75%的转录将被抑制。而核小体与组蛋白H1的交联会加大这一抑制程度[36]。通过进一步研究,科学家们推测,核小体组蛋白的静电位阻很可能是这种阻碍效应的重要原因。核小体组蛋白带正电荷,而DNA带负电荷,根据同性相斥,组蛋白的静电位阻作用排斥其他蛋白质分子与核小体DNA结合,从而阻断RNA聚合酶、转录因子、DNA复制与修复酶等与DNA的接触机会,抑制了遗传信息的表达。而NFR以及两核小体间的linker DNA由于不存在组蛋白的静电位阻效应,转录因子等可顺利与顺式调控元件结合,从而启动基因转录[37]。
2 染色质的二维构象
通过形成核小体,DNA的长度压缩了6~7倍,但这还远远达不到存储于直径仅为10 µm的细胞核中的程度。这意味着,染色质拥有比核小体更高级的构象类型。1979年,Finch等[4]最先通过电子显微镜观察到了直径为30 nm左右的染色质纤维,电镜结果提示该结构由11 nm核小体串珠结构组成。30 nm染色质纤维的发现正式使染色质构象研究推进到关注核小体间相互作用关系的二维层面。针对30 nm染色质纤维的结构模型,科学家们做了大量的工作,并提出了不同的猜想,如Solenoid模型[4,38]、Helical Ribbon模型[39]、two-start的Zigzag模型[40]等。2014年,通过冷冻电镜单颗粒三维重构技术,30 nm染色质纤维的高清晰三维结构被成功获取[41]。电镜结果显示,30 nm染色质纤维以4个核小体为结构单元;各单元之间通过相互扭曲折叠,从而形成一个左手双螺旋高级结构。其中,“四核小体”结构单元这一结果在染色质单分子力学研究过程中得到再次印证[42]。研究人员发现,“四核小体”结构是染色质折叠/去折叠过程中存在的一种稳定的中间状态,进而推测该结构单元可能是除核小体以外的,一个具有重要调控功能的结构与功能单元,可通过调控染色质纤维结构的紧密程度参与基因转录调控过程[42]。但事实上,以上所有关于30 nm染色质纤维的结果都是基于体外实验发现的,截止至今,科学家尚未真正在细胞内发现该结构的存在,因 此,关于染色质30 nm纤维的体内研究还有待进一步深入。
目前,关于染色质二维构象与遗传信息传递功能方面的研究相对稀少,更多的是一些推测和猜想。例如通过Hi-CO方法,Ohno等[43]首次发现了核小体在折叠过程中存在两种基本的结构单元:α-四面体(α-tetrahedron)和β-菱形(β-rhombus)。其中,核小体相对距离更远的β-菱形单元更倾向于分布在代表启动子的H2A.Z组蛋白附近,并与转录激活标记如H3K18ac和H3K4ac的富集呈正相关关系。而结合观测到的随核小体相对距离增加,基因表达显著上调这一结果,Ohno等[43]推测基因的激活与染色质的二维构象β-菱形单元相关。
3 染色质的三维构象
染色质三维构象的研究起始时间较早,早在1885年Rabl[44]就观察到细胞核内存在不同的染色体区域,而后通过荧光染色技术、显微技术等手段,很多实验都证实了细胞核中存在不同的三维结构[45]。但由于缺乏微观的证据,染色质三维构象并没有获得太多的关注。随着测序技术的发展,“人类基因组计划”(human genome project, HGP)[46]和“人类基因组百科全书计划”(encyclopedia of DNA elements, ENCODE)[47]的完成,科学家们逐渐意识到,基因组在空间结构上并不是一维线性排开的,早前的线性分子模型已不足以揭示这些新发现的离散的调控元件以及结构变异与基因功能的联系。至此,三维基因组学研究热潮被正式掀起。染色质构象捕获技术(chromosome conformation capture, 3C)是Dekker等[48]于2002年开发的测定特定的点对点之间染色质交互作用的新技术。该技术第一次将认识DNA一维序列高度提升到三维水平,也成为了后续三维基因组测序技术开发的基础。目前,基于3C技术衍生出的构象捕获技术包括4C[49,50]、5C[51]、Hi-C[6]、ChIA- PET[52]、原位In situ Hi-C[9]、高效酶切DNase Hi-C[53]、杂交探针Capture-Hi-C[54]、单细胞Hi-C[55]、DLO Hi-C[56]等,为研究基因间的远距离互作,诠释转录因子与染色质相互作用的关系,以及细胞核内互作染色质的空间构象提供了重要的基础。此外,随着电镜技术的发展,超高分辨率电子显微镜技术也为染色质三维构象的研究提供了新的视角。
基于上述技术和分析结果,科学家们提出了真核生物染色质三维层级假说模型。在不同的空间尺度上,这些层级结构依次为染色质疆域(chromosome territory, CT)[5]、染色质区室(chromatin compartment A/B)[6],拓扑相关结构域(topologically associating domain, TAD)[7,8]和染色质环(chromatin loop)[9](图2)。
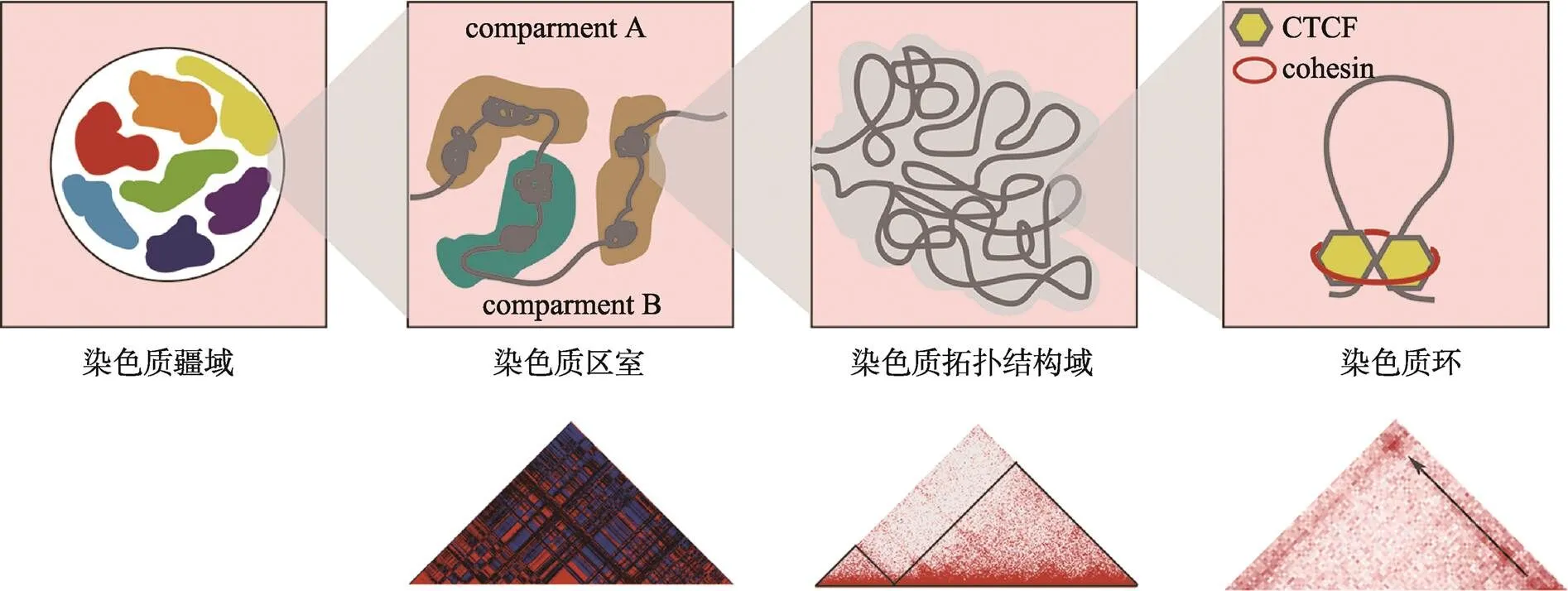
图2 染色质的三维构象
Compartment A/B及TAD热图数据来源于小鼠胚胎干细胞eHi-C (专利号CN201610995880.X)数据,chromatin loop热图数据来源于人类淋巴母细胞(GM12878)Hi-C数据[9]。
3.1 染色质疆域
在细胞核内,每条染色体的分布相对独立,它们各自占据着一块特定的、不重叠的区域,彼此间少有接触,而这个起着屏障作用的区域即是CT。大量研究显示,CT的径向定位具有偏好性,且在进化上高度保守。一般而言,小的、富含基因的染色体位于细胞核中心附近,而大的、缺乏基因的染色体位于细胞核边缘附近。Koehler等[57]通过对牛胚胎的研究发现,CT径向定位偏好性的首次出现与胚胎基因组的激活有关,并在囊胚中完全建立。此外,也有相关研究表明,CT的定位与细胞复制时间和转录活性相关。早期复制位点和活性基因倾向于在细胞核内部定位,而晚期复制位点和抑制基因则倾向于在核边缘定位[58,59]。这种现象与普遍认可的核纤层抑制基因表达的观点相符。但也存在例外,如夜行哺乳动物的视网膜杆状细胞中的CT定位与大多数白天活动的真核生物的相反,表现为异染色质定位于核中心,常染色质位于核外围[60]。
3.2 染色质区室
2009年, Lieberman-Aiden[6]首次运用Hi-C技术揭示了人淋巴母细胞的核内三维构象,并提出了染色质空间构象的另一重要特征,即染色质由compartment A和compartment B交替分布构成。Compartment A结构具有基因密集、转录活跃、可接近性强(DNase Ⅰ高度敏感)等特征,而compartment B则恰恰相反,该区域折叠程度高,基因分布少,且多富集具有抑制活性的组蛋白如H3K27me3等[6]。通过关联分析,科学家们发现,compartment A和compartment B分别与细胞学上的常染色质和异染色质相对应。而除了在基因表达特征层面,compartment B在细胞核内的分布也与异染色质十分相像,与核纤层结合结构域(nuclear lamina-associated domains, LADs)有较大的重叠,主要定位于核纤层附近[61]。这些证据在一定程度上说明了compartment A/B实质上是对常、异染色质具有不同亚细胞核定位的再发现。
Compartment A/B具有一定的细胞特异性,伴随着细胞状态的改变,可发生compartment A/B的互换。例如,在人胚胎干细胞分化过程中,加州大学圣地亚哥分校的Bing Ren团队[62]发现至少有36%的基因组发生了compartment A/B的转换。而结合RNA-seq数据,可清晰的观察到compartment B到A的转变,使基因更倾向于高表达,而compartment A到B的转变,基因表达则倾向于被抑制。该研究表明compartment A/B参与基因转录表达的调控过程,对基因的特异性表达有重要的影响作用。此外,相关研究人员还发现,compartment A/B与DNA的复制时序相关。Compartment A到compartment B的转变和早期到晚期的复制时序变化相对应,且在发生时间上一致,而compartment B至compartment A的改变则早于晚期到早期复制时间的变化和转录激活[63]。进一步将compartment A/B细分成类亚区室(subcompartment),Rao等[9]的研究结果显示,活跃的A1、A2 subcompartment DNA复制发生时间更早,在细胞周期S期更早期的阶段,而抑制状态的B1、B2和B3的复制时间则与S期的中后期对应。
3.3 染色质拓扑结构域
TAD是基因组上长度从几百kb到几Mb不等的DNA片段,其内部含有一个或多个基因和调控元件,可视为一种高度自我互作的基因组单元。相关研究表明,TAD的边界富集了绝缘子结合蛋白CTCF、持家基因、tRNA、短散点元件(SINE)、H3K4me3、H3K36me3和黏连蛋白复合物(cohesin complex)等,科学家们推测这些因子与TAD的形成相关[7]。TAD在不同物种、不同细胞类型以及不同进化阶段中具有高度的保守性[64,65]。如50%~70%的TAD同时存在于小鼠和人类胚胎干细胞中[62],而随着分化的进行,TAD的位置几乎不会发生变化[66]。因此,TAD被认为是染色质折叠的基本结构单元。
TAD与基因转录表达间的关系一直是科研界的研究热点。不同的研究表明,TAD与多种生物学现象间有着紧密的关联。如TAD边界与复制结构域高度吻合[67];在胚胎干细胞分化过程中,TAD经历了从无到有的变化过程[68];在小鼠细胞中,雌性X染色体的沉默伴随着TAD的重构[69,70];一些疾病的发生以及机体应激反应过程中伴随的TAD边界的移动或消失[71~74]。这些研究都在一定程度上暗示了,TAD可能是基因组发挥功能的一类基本单元,它们内部的协同调控及边界的屏障作用很可能是其发挥生物学功能的主要作用模式。
TAD内部的协同调控主要是指TAD将转录调控元件限定在同一个区域内,有效构建自主的基因调控域,独立调控该区域的基因表达(图3)。近年来,不少实验都为该观点提供了证据支持。如在不同细胞和组织中,同一TAD内部的基因表达均呈现出了趋同性,拥有更高的相关性[8,62,75]。而将报告基因随机插入到小鼠基因组中,Symmons等[76]发现报告基因的表达与同一TAD范围内基因的表达特征相仿。但也有科学家认为,这种作用其实是很微弱的,与可同时激活所有基因的操纵子不同,TAD的协同调控作用可能只影响单个TAD中的一个基因子集,在功能上具有协同效应的基因往往更倾向于位于同一个TAD内[8,77]。其中,协同影响调控元件活性的数量性状位点(quantitative trait loci, QTL)倾向于分布在同一个TAD中[78,79];相关基因簇被划入单个TAD中[80]即为有力佐证。当前,TAD内部协同调控作用模式为细胞中基因共表达现象做出了较好的解释。
内部互作频率高,组间互作频率低是TAD的一个基本特征,也是TAD屏障作用最直接的体现(图3)。由于大多数调控元件与启动子的空间交互受到TAD结构域的限制[81,82],科学家们推测TAD边界的保持对基因正确的时空表达具有重要的意义。目前,已有研究表明,TAD边界的破坏、移位都会造成基因表达的紊乱。例如敲除小鼠胚胎干细胞X染色体上的TAD边界,可明显观测到两个TAD间的互作增加,邻近TAD基因表达上调[8]。而基因所在TAD边界的敲除、倒位,可使基因的增强子分别错误地激活、和基因,发生异位表达,进而导致短指、多指畸形和F综合症的发生[71]。此外,一些报道指出,TAD边界在降低转录噪音上有一定的贡献。许多启动子和增强子具有双向转录特性,从而产生转录噪音,而在TAD边界处,这种异向转录可被有效阻止[83]。这些结果都暗示着TAD边界是阻止基因组内活动扩散的物理屏障,通过阻断不必要的互作,使基因发挥正常的作用。但在最近的一篇报道中,一个有趣的现象却打破了这种认识。通过人为敲除TAD边界和CTCF位点诱发TAD融合,Alexandra等[84]发现TAD边界的消失并没能引起基因显著的表达变化。这说明TAD“屏障”的建立可能并不仅仅依赖于边界这一单一因素,TAD结构调节基因表达的强度和准确度有待进一步探究。
3.4 染色质环
根据染色质纤维的聚合特性,由于随机碰撞,两个相距较远的基因组位点会以非常低的频率相互接触[6,85]。然而,某些位点的偶然接触频率却明显高于预期。科学家们将这种连接高频互作位点的染色质环状结构命名为loop。Loop由CTCF和cohesin等因子介导形成[86,87],通常发生在同一个TAD或sub-TAD中[82]。根据两端连接元件的不同,loop可细分为启动子–增强子loop、启动子–启动子loop、增强子–增强子loop等,这不同的loop交互作用,共同形成一个复杂的调控网络[81,88,89]。
Loop的形成与基因表达调控有着紧密的关联。其中,β-珠蛋白基因簇启动子与远端基因座控制区(locus control regions, LCR)的互作即为一个典型的案例。当β-珠蛋白基因簇与LCR形成染色质环时,基因表达被显著增强,反之,基因表达则处于一个极低水平[90,91]。而根据最新报道,美国丹娜-法伯癌症研究所Rani E. George课题组发现在治疗抵抗的癌细胞中,CTCF同源物BORIS调节的loop的变化可导致超级增强子(包含多个增强子,具有高转录因子密度,并能显著促进基因表达的基因组区域)的形成,从而驱动一类proneural转录因子的异位表达,最终产生抗ALK抑制的表型[92,93]。通过人为干预loop的形成与消失,越来越多的研究结果证明loop是基因转录发生变化的一个重要原因,而不是一个结果。如在转录因子GATA1的缺失的情况下,Deng等[91]通过将Lbd1加在一个锌指蛋白上,牵引该分子到达β珠蛋白启动子上的靶位点处,诱发形成一个位于增强子和启动子之间的染色质环,可使原本处于抑制状态的β-珠蛋白基因的表达获得明显的提高。而利用CRISPR/Cas9技术缺失前列腺癌风险区域,破坏该区域和互作形成的loop结构后,原本被抑制的表达显著上调,并成功激活诱导的致癌基因的表达[94]。从这些结果可推测,loop是基因激活与沉默依赖的介导因素,在DNA编码信息传递到RNA上的这个基础过程中起着不可或缺的关键作用。
在细胞分化过程中,loop具有两种作用模式,分别是指导模型(instructive model)和许可模型(permissive model)[95](图3)。指导模型认为,随着分化的推进,区域内转录因子的不同,导致细胞新形成具有组织特异性的loop,进而调控特定基因的表达。如α-珠蛋白基因和β-珠蛋白基因的增强子–启动子loop只存在于红细胞中,其互作频率在红细胞成熟过程中逐渐增强[96,97]。增强子与远端元件的互作只在高表达胸腺细胞中出现[98]。而许可模型则是指loop结构以沉默状态存在于祖细胞中,不同分化时期的转录因子选择性地将其激活,从而发挥特定的基因转录调控作用。如与增强子ZRS的loop结构同时被观测到出现在胚胎干细胞和下肢细胞中,但只有在下肢细胞中,才会表现为上调表达[7]。这两种模型在一定程度上解析了基因组织特异性表达的作用机理,也进一步暗示了loop是基因表达调控的必要不充分条件,具有重要的研究价值。
4 染色质构象的动态调控
不同的细胞表型特征,背后往往对应着特定的染色质构象;不同维度、不同状态的染色质构象对DNA复制、转录及修复等过程起着重要的调控、甚至是决定作用。但实际上,DNA的高度压缩、折叠对这些过程中各种蛋白、调控因子“读取”和“访问”遗传信息起到的却是阻碍作用。细胞如何打破这种阻碍,如何实现染色质构象的动态变化成为了科学家研究的一个热点。基于目前研究报道,染色质构象的动态调控模式主要有以下4种:DNA甲基化、ATP依赖的染色质重塑复合物、组蛋白修饰或组蛋白变体以及相分离(图4)。

图3 TAD与loop的作用模式
A:TAD作用模式;B:loop作用模式。
4.1 DNA甲基化
DNA甲基化是指在DNA甲基转移酶的作用下,将甲基基团转移到腺嘌呤或胞嘧啶碱基上的一种修饰方式,主要发生在富含双核苷酸CpG岛的区域[99,100]。CpG岛的高甲基化会导致基因的表达下降或沉默,而染色质构象的变化是其中的一个重要原因。研究显示,基因上非启动子区的DNA甲基化能减少甲基化区域的H3K4me2/me3、H3K9Ac和H3K14Ac的修饰形式,从而使染色质结构紧密化,减少在甲基化区域的RNA Pol II的结合,最终影响转录速率[101]。而单碱基稀疏保守低甲基化CpG (scUMC)增高可减弱染色质环因子结合DNA的强度,从而减少DNA环绑定结两端的互作,导致相应DNA环调控的基因表达下降[102]。一般而言,DNA甲基化会引起染色质的高度压缩,形成非活跃状态的异染色质,从而导致基因沉默,去甲基化则会使染色质结构松散,进而激活基因的转录表达[103]。Compartment A的甲基化水平显著低于Compartment B即是该观点的一个印证[13]。但也有研究结果表明,DNA甲基化所导致的染色质构象变化并非一定带来基因表达下调,Flavahan等[104]对携带异柠檬酸脱氢酶基因()突变的脑瘤进行研究时发现,TAD边界CTCF位点的甲基化,可使CTCF结合减少,进而打破TAD边界,使原本被TAD边界分隔的两个调控元件发生强烈互作,激活相关基因的表达。
4.2 ATP依赖的染色质重塑复合物
ATP依赖的染色质重塑复合物即是可利用ATP水解的能量来驱动核小体结构改变,从而改善转录因子等在染色质DNA局部的可接近性的蛋白因子。根据复合物中酶活中心蛋白亚基ATPase结构域相邻的其他结构域的组成,染色质重塑复合物被分为ISWI (imitation switch),SWI/SNF (mating type switching/sucrose non-fermenting),CHD (chromodomain helicase DNA-binding)和INO80 (inositol requiring 80)4个亚家族[105]。
介导核小体“滑动”和“置换”是ATP依赖的染色质重塑复合物发挥染色质重塑功能的主要方式。研究表明,ATP依赖的染色质重塑复合物具有类DNA移位酶作用,即在DNA双链未解开的情况下可以使核小体沿着DNA滑动[106]。目前,最为详尽的一个移动模型为陈柱成和李雪明课题组[107]在研究不同核苷酸状态下Snf2-核小体复合物的冷冻电镜结构时提出的两步走“DNA波”模型,即第一步,ATP水解,Snf2张开,把DNA从入口端拉进,并在Snf2与核小体的结合点(SHL2)处储存1 bp DNA形变(“DNA波”);第二步,ATP结合,Snf2关闭,使得DNA形变向出口端传递,就像水波沿湖面传递一样,最终实现DNA对组蛋白的相对移动。而该模型在关于ISWI驱动核小体滑移机制的研究中也得到了证实[108]。介导核小体“置换”是指重塑复合物通过核小体中组蛋白变异体与经典组蛋白之间的替换,从而改变染色质构象。其中最典型的例子就是在酵母中Swr1催化H2AZ-H2B异源二聚体与核小体中经典H2A-H2B二聚体之间的替换[109],但其具体机制还有待进一步研究。

图4 染色质构象动态调控机制
A:DNA甲基化;B:ATP依赖的染色质重塑复合物;C:组蛋白修饰;D:相分离。
4.3 组蛋白修饰或组蛋白变体对染色质结构的影响
组蛋白的N端拖尾伸出核小体外,可被共价修饰,进而改变染色质构象,导致转录的激活或抑制。目前,该类修饰主要包括乙酰化、甲基化、磷酸化、泛素化等[110]。组蛋白修饰影响染色质构象的方式主要有两种:一是影响核心组蛋白的电荷平衡,如组蛋白乙酰化后可中和其部分的正电荷,松弛组蛋白对DNA的结合,使染色质重构,以便转录因子与DNA结合,进而激活转录[111~113];二是影响组蛋白与其他蛋白相互结合的功能,如核心组蛋白H4的Lys16乙酰化阻止其与SIR3的相互作用,因而抑制异染色质的形成[114];而组蛋白H3 N端尾部的Lys9甲基化可吸引异染色质蛋白1(heterochromatin protein 1, HP1),使邻近组蛋白H3的Lys9被甲基化,从而导致异染色质区域的扩展[115]。
组蛋白变体是一类与经典的组蛋白序列高度相似,但功能却不同的变异体。目前,除H4外,H1、H2A、H2B和H3均有其相对应的组蛋白变体,它们通过与经典组蛋白发生置换,在染色质构象的动态调控中发挥着重要的作用[116]。例如,在胚胎干细胞中,H3.3 参与异染色质的形成[117]。同时也有研究表明,H2A.Z相比于H2A更能促进染色质高级结构的形成,并和异染色质蛋白HP1α协同作用维护组成型异染色质结构[118,119]。但对于组蛋白变体在转录调控过程中所发挥的功能及其机理至今还没有明确的解析。
4.4 相分离
相分离(phase separation)描述的是一种细胞里不同成分间相互碰撞、融合形成液滴,从而使一些成分被包裹在液滴内,一些成分被阻隔在液滴外的现象,它普遍存在细胞中,并与细胞结构的形成密切相关[120]。最新研究发现,相分离在染色质构象形成过程中具有突出贡献[121~123]。Strom等[121]在对早期果蝇胚胎和小鼠细胞进行研究时发现,相分离介导异染色质的形成,并提出了新异染色质形成模型。Strom等[121]认为HP1α蛋白发生液相分离,形成容纳染色体而排斥RNA聚合酶等分子的液相稳定区室,是异染色质形成的第一步。而该模型已获得体外实验、果蝇模型和人类细胞中的证据支持。为进一步研究相分离与染色质构象之间的关系,Shin等[124]开发了CasDrop体系,该体系通过人为调控相分离的发生,可直接观测相分离对染色质构象的影响。通过CasDrop,Shin等[124]发现相变易发生在染色质结构较为疏松、应变能较低的区域;染色质致密区域的较小的相变液滴则最终倾向于溶解;液滴对染色质的排斥会导致染色质结构的重塑。此外,Shin等[124]提出了染色质过滤模型(chromatin filter model),即结合在特定基因位点的液滴通过融合能够将距离较远的基因拉近,且排斥不含结合位点的背景基因区域,为研究染色质构象与基因转录表达关系提供了新的研究思路。但值得注意的是,相分离影响染色质构象方面的研究仍处于初期阶段,要真正揭示其中的关联必需更多的实验支持。
5 挑战与展望
随着近年来不同染色质构象捕获技术的兴起,染色质构象研究领域出现了前所未有的发展,为进一步解读生物遗传密码开辟了新的途径,但同时也面临着大量的挑战。目前,在实验上,基于高通量测序的三维基因组数据仍存在信噪比低、分辨率不佳、数据质量参差不齐等问题。在数据分析上,基于bin矩阵的算法占据主导地位,该方法通过互作信号的中点坐标,将限制性片段分配到相应区间,并通过bin-bin交互观察计算相应结构,其计算结果深受bin本身大小的影响,容易产生偏差;此外,由于相关分析算法各有倚重,缺乏统一的度量和评判标准,不同软件分析结果的可重复性让人担忧,如对于同一数据同一分析目的,HICUP、FitHiC和Homer获取loop的数目和假阳性率相差甚远[125];如此种种,不仅使不同的数据之间缺乏可比性,造成大量数据资源的浪费,更让由此建立起来的假说和理论不断的受到考验和挑战;如Eileen Finn等[126]在利用模式动物果蝇研究高度重排的突变染色体时发现,大多数TAD变化对基因的表达几乎没有影响。这种TAD变化与基因表达之间的“脱钩”现象彻底颠覆了人们对TAD调控基因表达的认识。
染色质构象研究是一个多学科交叉的研究领域。化学、物理、生物和计算机等多学科的参与为染色质构象的研究提供了不同的研究思路和角度,但与此同时也催生了一些新的问题,包括如何实现这些不同层面数据间的关联分析,去伪存真。目前,不同技术间的染色质构象分析结果差异较大,例如关于TAD的有无,高通量测序技术与超高分辨率ChromEMT电镜技术给出了截然相反的结果[127]。而尽管MERFISH超高分辨成像技术在单细胞层面观测到了类TAD结构域,其高度的异质性却与传统概念上的TAD的保守特征相悖,完全打破了人们对传统TAD的定义[128]。这到底是电镜技术缺陷使然,还是说TAD仅仅是个统计模型,并不存在于细胞核中,至今没有一个合理的解释。
染色质构象具有异质性,且时刻处于变化过程中,在单细胞水平捕捉到这种结构上的动态变化是染色质构象精确研究的基础也是目前面临的一大难题。截止目前,尚无一技术可实现高通量的、在未破碎的细胞即活细胞中直接观测到染色质构象变化。而标榜在单细胞水平捕捉染色质构象的单细胞Hi-C也因分辨率低下问题,无法捕捉到除compartment A/B以外的染色质高级构象[55]。这也就意味着,人们常说的染色质构象的动态变化实为群体细胞的“死”动态变化,并不能很好的揭示染色质构象的本质特征。如何在单细胞水平上实现高分辨率、高效、实时地监测染色质构象变化成为了一个亟待解决的问题。
揭示染色质构象与基因功能关系是染色质构象研究的最终目的,也是深入解读遗传密码关键的一步。染色质构象与基因功能的阐明要求多组学数据的联合分析,但目前同一样品多组学数据的同步分析实现起来还存在一定的难度。对于电镜下瞬时的构象特征,如何捕捉到与此相对应的转录组、蛋白组、代谢组、表观组的特征也成为了一个难题。由于细胞的特异性、取样时无法避免的时间误差,分别来源于不同细胞的多组学数据往往会导致多组学特征关联分析出现偏差。此外,针对多组学分析的成熟的算法亟待开发。目前,多组学数据的分析往往需要调用多个软件,设置十几个参数,过程纷繁复杂,各实验室流程各异,对后续的深入研究带来了极大的不便。
染色质构象变化是最基础的生物学现象,已成为联系不同生物学过程的重要桥梁和纽带。染色质构象研究的推进有赖于复制、转录、翻译等一系列生物学理论的完善,实验技术的改进,更需要多组学、多学科、多手段的有机整合。例如染色质构象捕获与基于免疫沉淀和tagmentation的文库制备相结合,可有效提高信噪比,在极少样本条件下,获取大量的构象信息[129];不同荧光分子和成像技术的引入,在提高分辨率的同时可在基因组尺度上实现其他分子如新生RNA行为的同步观测,为多组学数据的无缝整合提供了可能[130];大数据分析方法,如卷积神经网络[131]、自举式深度学习模型[132]等的应用可在一定程度上弥补分辨率不足的问题;而眺望未来,人工智能和深度学习算法的应用不仅能为特定的场景和特定的问题提供预测性模型和答案,更有可能一秒实现追本溯源,告诉我们生物学上到底发生了什么才会产生我们所看到的数据。我们有充分的理由相信,各领域的优势互补、不囿传统的大胆创新将成为推动“4D核体计划”[133]的强大动力,实现从空间(三维)和时间(第四维度)角度研究细胞核结构形成原理,进一步探究其对基因表达、细胞功能,以及对发育和疾病发生、发展的影响,揭示遗传信息最深层的奥秘。
[1] Kornberg RD. Chromatin structure: A repeating unit of histones and DNA., 1974, 184(4139): 868–871.
[2] Guertin MJ, Lis JT. Mechanisms by which transcription factors gain access to target sequence elements in chromatin., 2013, 23(2): 116–123.
[3] Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome., 2012, 489(7414): 75–82.
[4] Finch JT, Klug A. Solenoidal model for superstructure in chromatin., 1976, 73(6): 1897–1901.
[5] Boveri T. Die blastomerenkerne von ascaris megalocephala und die theorie der chromosomenindividualität., 1909, 3: 181–268.
[6] Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of Long-Range interactions reveals folding principles of the human genome., 2009, 326(5950): 289–293.
[7] Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions., 2012, 485(7398): 376–380.
[8] Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre., 2012, 485(7398): 381– 385.
[9] Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping., 2014, 159(7): 1665–1680.
[10] Zhang YB. 3D Genomics and precision biology., 2018, 34(4): 351–363.张玉波. 三维基因组学与精准生物学. 中国生物化学与分子生物学报, 2018, 34(4): 351–363.
[11] Risca VI, Greenleaf WJ. Unraveling the 3D genome: Genomics tools for multiscale exploration., 2015, 31(7): 357–372.
[12] Ning CY, He MN, Tang QZ, Zhu Q, Li MZ, Li DY. Advances in mammalian three-dimensional genome by using Hi-C technology approach., 2019, 41(3): 215–233.宁椿游, 何梦楠, 唐茜子, 朱庆, 李明洲, 李地艳. 基于Hi-C技术哺乳动物三维基因组研究进展. 遗传, 2019, 41(03): 215–233.
[13] Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, Yao X, Li F, Zhu W, Gao L, Chen H, Du Z, Xie W, Xu X, Huang X, Liu J. 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis., 2017, 170(2): 367–381.
[14] Barutcu AR, Lajoie BR, Mccord RP, Tye CE, Hong D, Messier TL, Browne G, van Wijnen AJ, Lian JB, Stein JL, Dekker J, Imbalzano AN, Stein GS. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells., 2015, 16(1): 214.
[15] Taberlay PC, Achinger-Kawecka J, Lun AT, Buske FA, Sabir K, Gould CM, Zotenko E, Bert SA, Giles KA, Bauer DC, Smyth GK, Stirzaker C, O'Donoghue SI, Clark SJ. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations., 2016, 26(6): 719–731.
[16] Brown KE. Chromatin folding and gene expression: New tools to reveal the spatial organization of genes., 2003, 11(5): 423–433.
[17] Almassalha LM, Tiwari A, Ruhoff PT, Stypula-Cyrus Y, Cherkezyan L, Matsuda H, Dela Cruz MA, Chandler JE, White C, Maneval C, Subramanian H, Szleifer I, Roy HK, Backman V. The global relationship between chromatin physical topology, fractal structure, and gene expression., 2017, 7: 41061.
[18] Hübner MR, Eckersley-Maslin MA, Spector DL. Chromatin organization and transcriptional regulation., 2013, 23(2): 89–95.
[19] Peng C, Li GL, Zhang YH, Ruan YJ. Reconstruction of three-dimensional structures of chromatin and its biological implications., 2014, 44(8): 794– 802.彭城, 李国亮, 张红雨, 阮一骏. 染色质三维结构重建及其生物学意义. 中国科学: 生命科学, 2014, 44(8): 794–802.
[20] Oudet P, Gross-Bellard M, Chambon P. Electron microscopic and biochemical evidence that chromatin structure is a repeating unit., 1975, 4(4): 281–300.
[21] Olins AL, Olins DE. Spheroid chromatin units (v bodies).. 1974, 183(4122): 330–332.
[22] Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 a resolution., 1997, 389(6648): 251–260.
[23] Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide., 2004, 36(8): 900–905.
[24] Sekinger EA, Moqtaderi Z, Struhl K. Intrinsic Histone- DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast., 2005, 18(6): 735–748.
[25] Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. Global nucleosome occupancy in yeast., 2004, 5(9): 1–11.
[26] Saragosti S, Moyne G, Yaniv M. Absence of nucleosomes in a fraction of SV40 chromatin between the origin of replication and the region coding for the late leader RNA., 1980, 20(1): 65–73.
[27] Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II., 2009, 325(5940): 626–628.
[28] Tilgner H, Nikolaou C, Althammer S, Sammeth M, Beato M, Valcárcel J, Guigó R. Nucleosome positioning as a determinant of exon recognition., 2009, 16(9): 996–1001.
[29] Schwartz S, Meshorer E, Ast G. Chromatin organization marks exon-intron structure., 2009, 16(9): 990–995.
[30] Mavrich TN, Jiang C, Ioshikhes IP, Li XY, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, Gilmour DS, Albert I, Pugh BF. Nucleosome organization in the Drosophila genome., 2008, 358(7193): 358–362.
[31] Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C. A high-resolution atlas of nucleosome occupancy in yeast., 2007, 39(10): 1235– 1244.
[32] Leimgruber E, Seguín-Estévez Q, Dunand-Sauthier I, Rybtsova N, Schmid CD, Ambrosini G, Bucher P, Reith W. Nucleosome eviction from MHC class II promoters controls positioning of the transcription start site., 2009, 37(8): 2514–2528.
[33] Segal E, Widom J. What controls nucleosome positions?, 2009, 25(8): 335–343.
[34] Schwartz S, Ast G. Chromatin density and splicing destiny: On the cross-talk between chromatin structure and splicing., 2014, 29(10): 1629–1636.
[35] Spies N, Nielsen CB, Padgett RA, Burge CB. Biased chromatin signatures around polyadenylation sites and exons., 2009, 36(2): 245–254.
[36] Laybourn PJ, Kadonaga JT. Role of nucleosomal cores and histone h1 in regulation of transcription by RNA polymerase II., 1991, 254(5029): 238–245.
[37] Jiang C, Pugh BF. Nucleosome positioning and gene regulation: Advances through genomics., 2009, 10(3): 161–172.
[38] Widom J, Klug A. Structure of the 3000Å chromatin filament: X-ray diffraction from oriented samples., 1985, 43(1): 207–213.
[39] Woodcock CL, Frado LL, Rattner JB. The higher-order structure of chromatin: Evidence for a helical ribbon arrangement., 1984, 99(1): 42–52.
[40] Benedetta D, Thomas S, Alexandra K, Sylwia D, Schroeder RR, Richmond TJ. Nucleosome arrays reveal the two-start organization of the chromatin fiber., 2004, 306(5701): 1571–1573.
[41] Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units., 2014, 344(6182): 376–380.
[42] Li W, Chen P, Yu J, Dong L, Liang D, Feng J, Yan J, Wang PY, Li Q, Zhang Z, Li M, Li G. FACT remodels the tetranucleosomal unit of chromatin fibers for gene transcription., 2016, 64(1): 120–133.
[43] Ohno M, Ando T, Priest DG, Kumar V, Yoshida Y, Taniguchi Y. Sub-nucleosomal genome structure reveals distinct nucleosome folding motifs., 2019, 176(3): 520–534.e25.
[44] Rabl C. Über zelltheilung., 1885, 10: 214– 330.
[45] Cremer T, Cremer M. Chromosome territories., 2010, 2(3): a003889.
[46] International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome., 2004, 431 (7011): 931–945.
[47] The ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA elements) project., 2004, 306(5696): 636–640.
[48] Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation., 2002, 295(5558): 1306–1311.
[49] Marieke S, Petra K, Erik S, Yuri M, Rob W, Elzo DW, Bas VS, Wouter DL. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C)., 2006, 38(11): 1348–1354.
[50] Zhao Z, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions., 2006, 38(11): 1341–1347.
[51] Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements., 2006, 16(10): 1299–1309.
[52] Fullwood MJ, Liu MH, Pan YF, Liu J, Han X, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, ChewEGY, Huang PYH, Welboren WJ, Han YY, Ooi HS, Ariyaratne PN, Vega VB, Luo AQ, Tan PY, Choy PY, Wansa KDSA, Zhao B, Lim KS, Leow SC, Yow JS, Joseph R, Li HX, Desai KV, Thomsen JS, Lee YK, Karuturi RKM, Herve T, Bourque BG, Stunnenberg HG, Ruan I, Cacheux-Rataboul V, Sung WK, Liu DT, Wei CL, Cheung E, Ruan YJ. An oestrogen receptor α-bound human chromatin interactome., 2009, 462(7269): 58–64.
[53] Ma W, Ay F, Lee C, Gulsoy G, Deng X, Cook S, Hesson J, Cavanaugh C, Ware CB, Krumm A, Shendure J, Blau CA, Disteche CM, Noble WS, Duan Z. Fine- scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes., 2015, 12(1): 71–78.
[54] Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, Eskiw CH, Luo YQ, Wei CL, Ruan YJ, Bieker JJ, Fraser P. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells., 2010, 42(1): 53–61.
[55] Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure., 2013, 502(7469): 59–64.
[56] Lin D, Hong P, Zhang S, Xu W, Jamal M, Yan K, Lei Y, Li L, Ruan Y, Fu ZF, Li G, Cao G. Digestion- ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture., 2018, 50(5): 754–763
[57] Koehler D, Zakhartchenko V, Froenicke L, Stone G, Stanyon R, Wolf E, Cremer T, Brero A. Changes of higher order chromatin arrangements during major genome activation in bovine preimplantation embryos., 2009, 315(12): 2053–2063.
[58] Cremer M, Grasser F, Lanctôt C, Müller S, Neusser M, Zinner R, Solovei I, Cremer T. Multicolor 3D fluorescence in situ hybridization for imaging interphase chromosomes., 2008, 463(463): 205– 239.
[59] Takizawa T, Meaburn KJ, Misteli T. The meaning of gene positioning., 2008, 135(1): 9–13.
[60] Solovei I, Kreysing M, Lanctôt C, Kösem S, Peichl L, Cremer T, Guck J, Joffe B. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution., 2009, 137(2): 356–368.
[61] Ryba T, Hiratani I, Lu JJ, Itoh M, Kulik M, Zhang JF, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types., 2010, 20(6): 761–770.
[62] Dixon JR, Inkyung J, Siddarth S, Yin S, Antosiewicz- Bourget JE, Ah Young L, Zhen Y, Audrey K, Nisha R, Wei X, Diao Y, Liang J, Zhao HM, Lobanenkov VV, Ecker JR, Thomson J, Ren B. Chromatin architecture reorganization during stem cell differentiation., 2015, 518(7539): 331–336.
[63] Miura H, Takahashi S, Poonperm R, Tanigawa A, Takebayashi S, Hiratani I. Single-cell DNA replication profiling identifies spatiotemporal developmental dynamics of chromosome organization., 2019, 51(9):1356–1368.
[64] Dekker J, Heard E. Structural and functional diversity of Topologically Associating Domains., 2015, 589(20PartA): 2877–2884.
[65] Dixon J, Gorkin D, Ren B. Chromatin domains: The unit of chromosome organization., 2016, 62(5): 668–680.
[66] Battulin N, Fishman VS, Mazur AM, Pomaznoy M, Khabarova AA, Afonnikov DA, Prokhortchouk EB, Serov OL. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach., 2015, 16(1): 77.
[67] Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, Thurman RE, Cheng Y, Gülsoy G, Dennis JH, Snyder MP, Stamatoyannopoulos JA, Taylor J, ardison RC, Kahveci T, Ren B, Gilbert DM. Topologically associating domains are stable units of replication-timing regulation., 2014, 515(7527): 402–405.
[68] Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, Yao X, Li F, Zhu W, Gao L, Chen H, Du Z, Xie W, Xu X, Huang X, Liu J. 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis., 2017, 170(2): 367–381.
[69] Wang CY, Jégu T, Chu HP, Oh HJ, Lee JT. SMCHD1 merges chromosome compartments and assists formation of Super-Structures on the inactive x., 2018, 174(2): 406–421.
[70] Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, Dekker J. Structural organization of the inactive X chromosome in the mouse., 2016, 535(7613): 575–579.
[71] Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions., 2015, 161(5): 1012–1025.
[72] Li W, Gong K, Li Q, Alber F, Zhou XJ. Hi-Corrector: A fast, scalable and memory-efficient package for normalizing large-scale Hi-C data., 2015, 31(6): 960–962.
[73] Lupiáñez DG, Spielmann M, Mundlos S. Breaking TADs: How alterations of chromatin domains result in disease., 2016, 32(4): 225–237.
[74] Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, Reddy J, Borges-Rivera D, Lee TI, Jaenisch R, Porteus MH, Dekker J, Young RA. Activation of proto-oncogenes by disruption of chromosome neighborhoods., 2016, 351(6280): 1454–1458.
[75] Zhan Y, Mariani L, Barozzi I, Schulz EG, Blüthgen N, Stadler M, Tiana G, Giorgetti L. Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes., 2017, 27(3): 479–490.
[76] Symmons O, Uslu VV, Tsujimura T, Ruf S, Nassari S, Schwarzer W, Ettwiller L, Spitz F. Functional and topological characteristics of mammalian regulatory domains., 2014, 24(3): 390–400.
[77] Yin S, Feng Y, Mccleary DF, Zhen Y, Lee E, Samantha K, Ulrich W, Jesse D, Leonard L, Lobanenkov VV, Ren B. A map of the cis-regulatory sequences in the mouse genome., 2012, 488(7409): 116–120.
[78] Grubert F, Zaugg JB, Kasowski M, Ursu O, Spacek DV, Martin AR, Greenside P, Srivas R, Phanstiel DH, Pekowska A, Heidari N, Euskirchen G, Huber W, Pritchard JK, Bustamante CD, Steinmetz LM, Kundaje A, Snyder M. Genetic control of chromatin states in humans involves local and distal chromosomal interactions., 2015, 162(5): 1051–1065.
[79] Waszak SM, Delaneau O, Gschwind AR, Kilpinen H, Raghav SK, Witwicki RM, Orioli A, Wiederkehr M, Panousis NI, Yurovsky A, Romano-Palumbo L, Planchon A, Bielser D, Padioleau I, Udin G, Thurnheer S, Hacker D, Hernandez N, Reymond A, Deplancke B, Dermitzakis ET. Population variation and genetic control of modular chromatin architecture in humans., 2015, 162(5): 1039–1050.
[80] Fritz AJ, Ghule PN, Boyd JR, Tye CE, Page NA, Hong D, Shirley DJ, Weinheimer AS, Barutcu AR, Gerrard DL, Frietze S, van Wijnen AJ, Zaidi SK, Imbalzano AN, Lian JB, Stein JL, Stein GS. Intranuclear and higher- order chromatin organization of the major histone gene cluster in breast cancer., 2018, 233(2): 278–1290.
[81] Sanyal A, Lajoie BR, Jian G, Dekker J. The long-range interaction landscape of gene promoters., 2012, 489(7414): 109–113.
[82] Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells., 2013, 503(7475): 290–294.
[83] Austenaa L, Barozzi I, Simonatto M, Masella S, Della Chiara G, Ghisletti S, Curina A, de Wit E, Bouwman BM, de Pretis S,Piccolo V, Termanini A, Prosperini E, Pelizzola M, de Laat W, Natoli G. Transcription of mammalian cis-regulatory elements is restrained by actively enforced early termination., 2015, 60(3): 460–474.
[84] Despang A, Schöpflin R, Franke M, Ali S, Jerković I, Paliou C, Chan WL, Timmermann B, Wittler L, Vingron M, Mundlos S, Ibrahim DM. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture., 2019, 51(8): 1263–1271.
[85] Bornfleth H, Edelmann P, Zink D, Cremer T, Cremer C. Quantitative motion analysis of subchromosomal foci in living cells using Four-Dimensional microscopy., 1999, 77(5): 2871–2886.
[86] Tang Z, Luo OJ, Nbsp O, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, Michalski P, Piecuch E, Wang P, Wang D, Tian SZ, Penrad-Mobayed M, Sachs LM, Ruan X, Wei CL, Liu ET, Wilczynski GM, Plewczynski D, Li G, Ruan Y. CTCF-Mediated human 3D genome architecture reveals chromatin topology for transcription., 2015, 163(7): 1611–1627.
[87] Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping., 2014, 159(7): 1665–1680.
[88] Zhang Y, Wong CH, Birnbaum RY, Li G, Favaro R, Ngan CY, Lim J, Tai E, Poh HM, Wong E, Mulawadi FH, Sung WK, Nicolis S, Ahituv N, Ruan Y, Wei CL. Chromatin connectivity maps reveal dynamic promoter- enhancer long-range associations., 2013, 504 (7479): 306–310.
[89] Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, Ong CT, Orlov YL, Hong S, Zhang Z, Landt S, Raha D, Euskirchen G, Wei CL, Ge W, Wang H, Davis C, Fisher-Aylor KI, Mortazavi A, Gerstein M, Gingeras T, Wold B, Sun Y, Fullwood MJ, Cheung E, Liu E, Sung WK, Snyder M, Ruan Y. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation., 2012, 148(1): 84–98.
[90] Deng W, Rupon JW, Krivega I, Breda L, Motta I, Jahn KS, Reik A, Gregory PD, Rivella S, Dean A, Blobel GA. Reactivation of developmentally silenced globin genes by forced chromatin looping., 2014, 158(4): 849–860.
[91] Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor., 2012, 149(6): 1233–1244.
[92] Debruyne DN, Dries R, Sengupta S, Seruggia D, Gao Y, Sharma B, Huang H, Moreau L, Mclane M, Day DS, Marco E, Chen T, Gray NS, Wong KK, Orkin SH, Yuan GC, Young RA, George RE. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells., 2019, 572(7771): 676–680.
[93] Peng YL, Zhang YB. Enhancer and super-enhancer: Positive regulators in gene transcription., 2018, 1(03): 169–179.
[94] Luo Z, Rhie SK, Lay FD, Farnham PJ. A prostate cancer risk element functions as a repressive loop that regulates HOXA13., 2017, 21(6): 1411–1417.
[95] de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes., 2013, 502(7472): 499–506.
[96] Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, De Laat W. The β-globin nuclear compartment in development and erythroid differentiation., 2003, 35(2): 190–194.
[97] Vernimmen D, De Gobbi M, Sloane-Stanley JA, Wood WG, Higgs DR. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression., 2014, 26(8): 2041– 2051.
[98] van De Werken HJ, Landan G, Holwerda SJ, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Öz Y, Bouwman BAM, Verstegen MJAM, De Wit E, Tanay A, De Laat W. Robust 4C-seq data analysis to screen for regulatory DNA interactions., 2012, 9(10): 969–972.
[99] Cooper DN, Krawczak M. Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes., 1989, 83(2): 181–188.
[100] Deng DJ. DNA methylation and demethylation: current status and future per-spective., 2014, 36(5): 403–410.邓大君. DNA甲基化和去甲基化的研究现状及思考. 遗传, 2014, 36(5): 403–410.
[101] Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells., 2004, 11(11): 1068–1075.
[102] Lin XQ, Su JZ, Chen KF, Rodriguez B, Li W. Sparse conserved under-methylated CpGs are associated with high-order chromatin structure., 2017, 18(1): 163.
[103] Gudjonsson JE, Krueger G. A role for epigenetics in psoriasis: methylated cytosine-guanine sites differentiate lesional from nonlesional skin and from normal skin., 2012, 132(1): 506–508.
[104] Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE. Insulator dysfunction and oncogene activation inmutant gliomas., 2016, 529(7584): 110–114.
[105] Witkowski L, Foulkes WD. In Brief: Picturing the complex world of chromatin remodelling families., 2016, 237(4): 403–406.
[106] Gangaraju VK, Bartholomew B. Mechanisms of ATP dependent chromatin remodeling., 2007, 618(1–2): 3–17.
[107] Li MJ, Xia X, Tian YY, Jia Q, Liu XY, Lu Y, Li M, Li XM, Chen ZC. Mechanism of DNA translocation underlying chromatin remodelling by Snf2., 2019, 567(7748): 409–413.
[108] Yan LJ, Wu H, Li XM, Gao N, Chen ZC. Structures of the ISWI-nucleosome complex reveal a conserved mechanism of chromatin remodeling., 2019, 26: 258–266.
[109] Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex., 2004, 303(5656): 343–348.
[110] Mersfelder EL, Parthun MR. The tale beyond the tail: Histone core domain modifications and the regulation of chromatin structure., 2006, 34(9): 2653–2662.
[111] Zhang R, Erler J, Langowski J. Histone acetylation regulates chromatin accessibility: Role of H4K16 in inter-nucleosome interaction., 2017, 112(3): 450–459.
[112] Wang C, Fu M, Pestell RG. Histone acetylation/ deacetylation as a regulator of cell cycle gene expression., 2004, 241(241): 207–216.
[113] Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code., 2005, 102(15): 5501–5506.
[114] Carmen AA, Milne L, Grunstein M. Acetylation of the yeast histone H4 N terminus regulates its binding to heterochromatin protein SIR3., 2002, 277(7): 4778–4781.
[115] Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9., 2002, 416(6876): 103–107.
[116] Zhao YQ, Jordan IK, Lunyak VV. Epigenetics componentsof aging in the central nervous system., 2013, 10(4): 647–663.
[117] Banaszynski LA, Wen D, Dewell S, Whitcomb SJ, Lin M, Diaz N, Elsässer SJ, Chapgier A, Goldberg AD, Canaani E, Rafii S, Zheng D, Allis CD. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells., 2013, 155(1): 107–120.
[118] Fan JY, Gordon F, Luger K, Hansen JC, Tremethick DJ. The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states., 2002, 9(3): 172–176.
[119] Fan JY, Rangasamy D, Luger K, Tremethick DJ. H2A.Z alters the nucleosome surface to promote HP1alpha- mediated chromatin fiber folding., 2004, 16(4): 655–661.
[120] Feng Z, Chen XD, Zhang MD, Zhang MJ. Formation of biological condensates via phase separation: Characteristics, analytical methods, and physiological implications., 2019, (40), doi: 10.1074/jbc.rev119. 007895.
[121] Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH. Phase separation drives heterochromatin domain formation., 2017, 547(7662): 241–245.
[122] Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, Narlikar GJ. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin., 2017, 547(7662): 236–240.
[123] Larson AG, Narlikar GJ. The role of phase separation in heterochromatin formation, function, and regulation., 2018, 57(17): 2540–2548.
[124] Shin Y, Chang Y, Lee DSW, Berry J, Sanders DW, Ronceray P, Wingreen NS, Haataja M, Brangwynne CP. Liquid nuclear condensates mechanically sense and restructure the genome., 2019, 175(6): 1481–1491.
[125] Forcato M, Nicoletti C, Pal K, Livi CM, Ferrari F, Bicciato S. Comparison of computational methods for Hi-C data analysis., 2017, 14(7): 679–685.
[126] Ghavi-Helm Y, Jankowski A, Meiers S, Viales RR, Korbel JO, Furlong EEM. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression., 2019, 51: 1272–1282.
[127] Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, O’Shea CC. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells., 2017, 357(6349): eaag0025.
[128] Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang XW. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells., 2018, 362(6413): u1783.
[129] Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture., 2016, 13(11): 919–922.
[130] Mateo LJ, Murphy SE, Hafner A, Cinquini IS, Walker CA, Boettiger AN. Visualizing DNA folding and RNA in embryos at single-cell resolution., 2019, 568(7750): 49–54.
[131] Zhang Y, An L, Xu J, Zhang B, Zheng WJ, Hu M, Tang JJ, Yue F. Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus., 2018, 9(1): 750.
[132] Li WR, Wong WH, Jiang R. DeepTACT: Predicting 3D chromatin contacts via bootstrapping deep learning., 2019, 47(10): e60.
[133] Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, Politz JCR, Shendure J, Zhong S. The 4D nucleome project., 2017, 549(7671): 219–226.
Linking chromatin conformation to gene function
Qitong Huang, Qing Li, Yubo Zhang
DNA is highly compressed and packaged as chromatin within the nucleus in eukaryotes. DNA replication, DNA repair and transcription, and other biological processes are precisely regulated or determined by chromatin conformation. Activation or repression of different genes/transcription factors are tightly correlated to their chromatin conformation which contributes to cell self-maintenance, differentiation, specificity and identity. Therefore, the study of bridging chromatin conformation to gene function has became crucial for decoding genetic information and precision biology which is also a great challenge for current genomics research. Here, we review the multiple aspects of chromatin conformation, including its history, characteristics, dynamics and the impact on different dimension-chromatin architecture to gene function. Additionally, we discuss the limitation and challenges of current status of linking chromatin conformation to gene function study. We believe this is informative and foundational in combing the context between chromatin conformation and gene function as reference.
chromatin conformation; gene function; dynamic regulation
2019-08-29;
2019-10-29
中国农业科学院科技创新工程、千人计划“青年项目”、中央级公益性科研院所基本科研业务费专项(编号:Y2019PT18-02)、国家自然科学基金面上项目(编号:31970592)和国家重点研发计划专项2018年度项目(编号:2018YFA0903201)资助 [Supported by the Agricultural Science and Technology Innovation Program, the Thousand Talents Plan for Young Professionals, the Fundamental Research Funds for Central Non-profit Scientific Institution (No.Y2019PT18-02), the National Natural Science Foundation of China (No.31970592) and the National Key R&D Program of China (No. 2018YFA0903201)]
黄其通,博士研究生,研究方向:动物三维基因组学。E-mail: miraclelive@qq.com
李清,硕士,研究方向:动物三维基因组学。E-mail: liqing9102@163.com
黄其通和李清并列第一作者。
张玉波,博士,研究员,研究方向:动物三维基因组学。E-mail: ribon_001@163.com
10.16288/j.yczz.19-257
2019/12/19 14:29:56
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20191218.1615.002.html
(责任编委:吴强)
