基于生物信息学的Hi-C研究现状与发展趋势
2020-03-04吕红强郝乐乐刘二虎吴志芳韩九强刘源
吕红强,郝乐乐,刘二虎,吴志芳,韩九强,刘源
综述
基于生物信息学的Hi-C研究现状与发展趋势
吕红强,郝乐乐,刘二虎,吴志芳,韩九强,刘源
西安交通大学电子与信息工程学院,西安 710049
染色体的空间交互作用被视为影响基因表达调控的重要因素,高通量染色体构象捕获(high-throughput chromosome conformation capture, Hi-C)技术已成为3D基因组学中探索染色体空间交互作用的主要实验手段之一。随着Hi-C样本数据的持续累积以及分析处理流程复杂度的不断提升,基于生物信息学的Hi-C数据分析对探究基因表达的时空调控机制而言,是机遇也是挑战。本文从生物信息学角度,综合阐述了Hi-C的国内外研究现状及发展动态,包括数据标准化、多级结构分析、数据可视化以及三维建模,重点剖析了多级结构中的A/B区室(A/B compartments)、拓扑相关域(topological associated domains, TADs)和染色质环(chromain looping),在此基础上分析了该方向未来可能的研究热点及发展趋势,以期为将基因表达调控的探索从传统线性空间进一步拓展到三维结构空间提供支持。
3D基因组学;Hi-C;生物信息学
基因表达的调控机制是现代分子生物学研究中的重要内容。其所研究的表达调控作用并不局限于传统的以染色体坐标为度量的一维线性结构,染色体的多级空间结构可使在线性坐标空间中的远程调控元件在三维结构空间中近距离调控目标基因的表达水平,因此染色体上各位点在细胞核中的空间交互作用被视为影响基因表达调控的重要因素。随着生物信息学领域相关研究的不断深入,高通量染色体构象捕获(high-throughput chromosome conformation capture, Hi-C)[1]技术逐渐成为探索染色体空间交互作用的主要技术手段[2],以此为核心的3D基因组学被称为基因组学研究的第三次浪潮[3]。当前,以Hi-C为代表的染色体构象捕获(chromosome conformation capture, 3C)[4]技术通过消化和重连空间上接近的染色体片段来确定不同位点之间的空间交互,为分析染色体在细胞核中的空间组织结构提供了有效途径。区别于早期3C技术的单点对单点检测,4C (chromosome conformation capture-on-chip)[5]技术的单点对多点检测,以及5C (chromosome conformation capture carbon copy)[6]技术的多点对多点检测,Hi-C将高通量测序技术与3C技术相结合,通过全点对全点检测,构建出全基因组范围内无偏的空间交互图谱[7]。正是由于Hi-C技术的这一优越性,才使得研究全基因组范围内的染色体三维结构成为可能[8]。
Hi-C技术通过消化和重连空间上接近的染色体片段,对其进行高通量测序,可确定染色体不同位点之间的空间交互作用。其生物实验的主要步骤包括:(1)交联,用甲醛对细胞进行瞬间固定,使DNA与蛋白,蛋白与蛋白之间相互交联;(2)酶切,利用限制性内切酶(如d Ⅲ)对DNA进行切割,使交联两侧产生粘性末端;(3)标记,修复切割末端,并用生物素标记末端;(4)连接,使用DNA连接酶通过平端连接切割末端生成嵌合分子;(5)解交联,对纯化后的DNA嵌合分子进行超声破碎或者利用限制性酶(Ⅰ)进行打断处理,筛选出被生物素标记的DNA片段,获得DNA文库;(6)测序,对DNA文库进行高通量双端测序。近年来,随着Hi-C技术的不断成熟,逐渐发展出系列Hi-C衍生技术,如promoter CHi-C[9]、single-cell Hi-C[10]、BL-Hi-C[11]和DLO Hi-C[12]等。相比于传统的Hi-C技术,promoter CHi-C利用RNA做诱饵筛选出包含启动子的DNA片段,可用于三维结构空间中启动子调控作用的分析;single-cell Hi-C技术用以单细胞染色体构象捕获,使得单细胞水平进行染色体空间交互作用的分析成为可能;BL-Hi-C通过对酶切和连接两个步骤的改进,具备高效和灵敏的结构性和调控性染色体构象捕获能力;DLO Hi-C则通过双交联和免生物素标记的方式,在简化流程的同时,有效降低了实验数据的背景噪音。
Hi-C生物实验产生数以亿计的配对末端序列片段(paired-end sequencing reads),这些两两配对的基因组序列片段,是染色体复杂空间结构在基因组片段水平上两两交互的分解,一般通过二维接触矩阵(contact matrix)的数据组织形式进行可视化和生物信息学分析处理。其中,根据实验数据所生成的二维接触矩阵即Hi-C接触矩阵也称其为交互矩阵,矩阵的行或列代表染色体坐标上固定长度的间隔区间,区间长度被称为分辨率,其值越小,分辨率越高,矩阵元素为落入相应行和列交互区间的配对末端序列片段的数量,称为交互频率(interaction frequency, IF),其值随着行和列之间距离的增加呈指数衰减。不同分辨率下的接触矩阵如图1所示。得益于Hi-C生物实验数据的快速增长,在Gene Expression Omnibus (GEO)[13]和Encyclopedia of DNA Elements (ENCODE)[14]等综合生物数据库以及Juicer[15]等Hi-C专业数据库中,已累积了大量的覆盖多个物种不同细胞系的Hi-C重复性样本数据。
随着Hi-C技术的不断成熟以及染色体各级空间结构的陆续发现,Hi-C数据的分析与处理已成为3D基因组学的研究热点之一。近年来,国内外已有多位专家学者对Hi-C方向的研究进展先后进行了综合性阐述,包括从3C到Hi-C的技术与方法的演进[3,16~18]、基于Hi-C技术的染色体多级结构[19]以及用于Hi-C数据分析的方法与工具进展[20]等。本文从生物信息学角度介绍了Hi-C的最新研究现状及发展动态,包括数据标准化、多级结构分析、数据可视化以及三维建模。在此基础上,分析了该方向未来可能的研究热点及发展趋势,以期成为现有Hi-C综述性成果在生物信息学方向的更新与补充,进而为将基因表达调控的探索从传统线性空间进一步拓展到三维结构空间提供支持。

图1 Hi-C接触矩阵
数据来源于Juicer数据集。
1 Hi-C国内外研究现状
1.1 数据标准化
Hi-C数据标准化用以移除生物实验过程中由各种不可避免的非随机因素所引入的样本间的系统偏差,是后续分析处理的数据质量前提。近年来,诸多Hi-C数据标准化方法陆续被提出。2011年,Yaffe等[21]提出一种基于集成概率模型的标准化方法,其通过序列片段长度、GC含量和序列映射得到先验概率,采用最大似然估计法确定模型参数。2012年,Cournac等[22]提出序列性组件标准化(sequential component normalization, SCN)方法,通过对单染色体接触矩阵的行列归一化产生标准化的双随机矩阵。2012年,Hu等[23]提出基于泊松回归模型的HiCNorm方法,在考虑序列片段长度、GC含量和序列映射3种因素的情况下,将回归后的残差作为标准化后的接触矩阵。2012年,Imakaev等[24]提出了面向全基因组的迭代修正和特征向量分解(iterative correction and eigenvector decomposition, ICE)方法,基于交互频率库规模等量和偏差分解思想进行接触矩阵的快速标准化。2013年,Knight等[25]提出一种矩阵平衡的数学方法(knight-ruiz,KR),后被广泛应用于Hi-C接触矩阵的标准化当中。2016年,Wu等[26]提出一种通过移除拷贝数偏差(copy number bias)对原ICE标准化进行改进的caICB方法。2018年,Stansfield等[27,28]提出基于局部加权线性回归的双样本标准化方法HiCcompare,并在2019年将其升级为有能力处理多组重复性样本的MultiHiCcompare方法。2019年,Spill等[29]提出基于负二项回归模型的Binless方法,其不依赖于接触矩阵分辨率,可在配对末端序列片段水平上进行Hi-C数据标准化。各主要Hi-C数据标准化方法如表1所示。目前,除Binless之外,Hi-C数据的标准化均是在接触矩阵水平上展开。接触矩阵上的标准化方法按照是否考虑系统偏差的具体来源类型可分为显式和隐式标准化,前者如HiCNorm和caICB,后者如SCN、ICE、KR、HiCcompare和MultiHiCcompare,其按照各样本间是否存在数据交互又可分为单样本和跨样本标准化,前者如SCN、HiCNorm、ICE、KR和caICB,后者如HiCcompare和MultiHiCcompare。

表1 Hi-C数据标准化方法
1.2 多级结构分析
染色体的构象具有多个层级结构[30],其结构单元由大到小依次为染色体疆域(chromosome territories)、A/B区室(A/B compartments)、拓扑相关域(topological associated domains, TADs)和染色质环(chromain looping)等(图2)。这些分级结构及其在基因表达调控中的作用,是目前Hi-C生物信息学分析的核心内容。通过对层级结构的鉴别可将模式复杂的交互作用矩阵转化为易于解读的特征信号,既便于样本间的比较,也便于与其他生物特征关联分析[19]。在此关注除染色体疆域(图2A)之外的分级结构。
1.2.1 A/B区室
A/B区室代表开放和关闭两种不同状态的染色体区域,A区室富含转录因子结合位点和活性组蛋白标记,属于转录活跃区域,而B区室含有抑制性组蛋白标记,属于转录抑制区域。2009年,Lieberman- Aiden等[1]在首次建立Hi-C技术的同时,利用特定距离上全基因组范围内的平均交互概率因子,对接触矩阵进行标准化,计算出行或列之间的皮尔逊相关系数矩阵,此矩阵的热图呈现出深浅交替的格子状模式(图2B),显示出两种不同结构特性的染色质状态,即A/B区室,通过对矩阵的主成分分析,发现第一主成分中的正负值区间信息分别对应A/B区室,其数值与基因密度、转录因子结合位点以及组蛋白标记等密切相关。2015年,Fortin等[31]提出通过不同类别的表观遗传数据,包括DNA甲基化微阵列、DNase超敏区序列、单细胞ATAC序列和单细胞全基因组亚硫酸氢盐序列等,预测多个细胞系下染色体A/B区室的方法,验证了A/B区室的结构和功能特性。2017年,山东农业大学农学院作物生物学国家重点实验室李平华实验室[32]发现大型植物的染色体可进一步划分为局部的A/B区室,这些区室反映了它们的常染色质、异染色质和多梳结构。在A/B区室识别方法研究及其结构特性分析的基础上,A/B区室与基因表达之间的关系也受到研究者的关注。2018年,Miura等[33]通过对Hi-C接触矩阵的主成分分析生成常染色体和X染色体上的A/B区室图谱数据,在此基础上,进一步分析了A/B区室的空间结构特征,并指出A/B区室的结构特异性及其与不同类型细胞中基因表达模式之间的联系。
1.2.2 拓扑相关域
拓扑相关域TADs是染色体区域内部交互作用水平远高于相邻区域的染色体结构单元,呈嵌套式(domain-in-domain)层级结构(图2C),已被证实广泛存在于真核生物的染色体当中[34,35]。TADs边界富集染色质调控蛋白CTCF、多种组蛋白修饰和持家基因等,其结构内部的基因持有共同的调控元件,如启动子和增强子等,这些基因在多种细胞系中存在协同表达特征,由此形成一个相对独立的调控单元,被认为是复制时间调控(replication-timing regulation)的稳定结构[34,36,37]。因此,TADs是染色体三维结构中的重要高阶结构单元和基因调控单元,对TADs的识别分析有助于理解染色体的复杂结构及其与生物学功能之间的关系。2012年,Dixon等[34]在最先发现接触矩阵中TADs结构的同时,提出一种互作方向指数(directionality index, DI)识别TAD边界点,并首次分析了TADs边界点附近CTCF和组蛋白修饰的高富集度以及基因的高表达水平特征。2014年,Levy-Leduc等[38]提出采用标准动态规划法,迭代求解TADs边界分割模型以得到TADs边界点的HiCseg方法。2015年,上海交通大学Shi联合美国南加州大学Shin等[39],提出TADs边界点识别方法TopDom,其采用钻石形滑动窗口法,提取接触矩阵对角线附近窗口内交互频率的统计特征,将特征曲线的局部极大值作为TADs边界点。2016年,Weinreb等[40]提出基于TADs内部交互频率的经验分布,进行层级式TADs识别的TADtree方法。2017年,Serra等[41]提出采用基于BIC惩罚的最大似然估计求解接触矩阵交互频率的概率模型,识别TADs边界点的TADbit方法。2017年,华中农业大学彭城等[42]提出层级式TADs识别方法HiTAD,其采用基于适应性交互方向指数的隐马尔科夫模型预测TADs边界点,在此基础上,采用迭代最优化策略搜寻接触矩阵中的层级式TADs。2017年,Haddad等[43]提出采用接触矩阵行或列的层次聚类,识别层级式TADs的IC-Finder方法。2017年,Yu等[44]提出采用高斯混合模型,进行层级式TADs识别的GMAP方法。2018年,Norton等[45]提出基于图理论进行层级式TADs识别的3DNetMod方法。2018年,中国科学院北京基因组研究所张治华团队联合北京航空航天大学计算机科学学院软件开发环境国家重点实验室李昂升团队,提出一种基于图结构熵理论的快速层级式TAD识别方法deDoc[46]。2018年,清华大学生物信息学教育部重点实验室陈阳等、南方科技大学前沿与交叉科学研究院李贵鹏等以及美国德克萨斯大学Zhang等[47],提出结合局部相对隔绝指数和多尺度聚类法进行TADs边界点识别的HiCDB方法。各主要TADs识别方法如表2所示。目前,除HiCDB之外,其他方法均不具备不同条件下TADs边界点差异性分析的能力。各方法按照是否考虑TADs的层级式结构又可分为边界点式和层级式两大类,前者如DI、HiCseg、TopDom和TADbit,后者如TADtree、HiTAD、IC-Finder、GMAP和3DNetMod。

图2 染色体多级结构
A:染色质疆域;B:A/B区室;C:拓扑相关域;D:染色质环。数据来源于Juicer数据集。

表2 TADs识别方法
1.2.3 染色质环
染色质环(chromatin loops)也可称为交互峰(interaction peaks),由染色体上相距较远的两个片段构成,其在线性空间中虽相距较远,但在三维空间结构中却具有显著的近距交互作用(图2D)。染色质环对理解染色体结构以及基因表达调控具有重要意义。2009年,Sexton等[48]基于3C技术研究了染色体的空间结构及其在基因表达调控中的作用,在分析染色体显著性交互作用的基础上,提出染色质环概念。2013年,复旦大学遗传工程国家重点实验室田卫东团队将Hi-C染色体空间交互数据引入到人类基因组作用元件与目标基因之间关系的预测当中,结果分析表明,基于Hi-C的染色质环信息能有效提升预测结果的生物功能特性及疾病相关性[49]。随后,染色质环的识别方法不断涌现。2014年,Ay等[50]对Hi-C数据中的随机聚合环和技术型系统偏差进行联合建模分析,提出了染色质环的识别方法Fit-Hi-C。2014年,Rao等[51]基于泊松分布模型提出了HiCCUPS方法,在去除TAD结构影响的前提下预测了染色质交互作用。2014年,Hwang等[52]基于负二项分布概率模型提出一种染色质环识别方法HIPPIE。2015年,Lun等[53]提出包括Hi-C配对末端序列片段预处理,数据标准化以及染色质环识别与差异分析的方法包dffHiC。2017年,中国科学院北京基因组研究所张治华[54]团队针对当时因Kbp分辨率Hi-C数据制备成本高昂而造成染色质环精确识别困难的问题,提出一种结合Kbp分辨率MNase- seq数据和低分辨Hi-C数据的染色质环精确识别方法CISD_loop。2018年,Djekidel等[55]基于空间泊松分布模型提出了检测染色质差异交互作用的方法FIND。随着对染色质环结构的深入了解,国内外相关学者也针对染色质环与病理之间的关系展开研究。2018年,Manduchi等[56]借助功能基因组学数据,分析了二型糖尿病患者基因组中增强子与启动子之间的空间交互及其与基因表达调控之间的关系,证实了增强子–启动子环在该类疾病发生发展中的作用。主要的染色质环识别方法如表3所示。按照针对显著交互作用还是差异交互作用进行鉴别可以划分为两种类型。其中,针对显著交互的有Fit-HiC、HiCCUPS、HIPPIE和CISD_loop,针对差异交互的有DiffiHiC和FIND。
1.3 数据可视化
数据可视化即为Hi-C数据的图形化显示及分析,最初的形式仅为接触矩阵的热图,随着Hi-C数据的不断累积及其分析处理复杂度的不断提升,一些Hi-C可视化平台相继出现。2013年,Zhou等[57]对原有Web Server服务器WashU Epigenome Browser进行升级,在已有不同物种不同组织与细胞系的表观基因组数据和转录组数据基础上,增添了远距基因组交互数据,其可借助三角形热图和两点间弧线对Hi-C和ChIA-PET数据中的空间结构关系进行图形化注解分析。2015年,Akdemir等[58]开发出一款Hi-C专用对比分析工具HiCPlotter,其将不同条件下的Hi-C矩阵热图与多能性因子、长非编码RNA以及结构蛋白等进行图形化并置,极大方便了基于Hi-C技术的染色体结构与功能对比分析。2016年,北京大学生命科学学院李程等[59]开发了一种Web Server服务器3Disease Browser,其实现了Hi-C数据、Chip-seq数据以及疾病相关染色体重排(chromosomal rearrangement, CR)数据的整合与可视化,具备对染色体特定重排区域进行三维立体可视化的能力。同年,Durand等[60]开发了基于云平台的Hi-C可视化软件Juicebox,该软件提供对外数据接口,支持染色体、分辨率和标准化方法选择、热图缩放以及与CTCF和RNA-seq等数据的关联分析等。2017年,Djekidel等[61]开发出的Web Server服务器HiC-3Dviewer,能够在三维空间中对Hi-C接触矩阵映射到染色体的相应区域进行交互式立体可视化,且具备ChIP-Seq和SNP数据标注功能。2017年,中国科学院北京基因组研究所张治华团队[62]开发出的Web Server服务器Delta,实现了Hi-C数据和ChIA-PET数据的可视化及结构分析,包括数据的交互式可视化、TADs和染色质环的结构预测以及基因组的三维建模。2018年,Calandrelli等[63]给出了开源的Hi-C可视化软件工具GITAR,该软件支持Hi-C数据预处理、标准化、TADs分析以及不同样本对比的可视化操作及结果显示。2018年,Wang等[64]开发的三维基因组Web Server服务器3D Genome browser,囊括了人类与小鼠的300多项不同类型数据,包括Hi-C、ChIA-PET、Capture Hi-C、PLAC-Seq、HiChIP、GAM和SPRITE,集成了包括ICE标准化、A/B区室识别和TADs识别工具的分析结果。同年,Wolff等[65]开发的集Hi-C数据预处理、接触矩阵标准化、A/B区室和TADs识别以及基因表达谱数据和Chip-seq数据等辅助分析于一体的可视化Web Server服务器Galaxy HiCExplorer,实现了Hi-C数据分析处理过程中绝大多数流程的数据可视化,人机交互更为友善。各主要Hi-C可视化工具软件如表4所示。

表3 染色质环识别方法
1.4 三维建模
三维建模是Hi-C的一项重要应用,其通过Hi-C数据的建模分析得到染色体的结构信息,从而在三维立体空间中重现染色体的物理结构,以辅助科学研究。2002年,Dekker等[4]在提出3C技术以及交互频率矩阵概念的基础上,借助聚合体柔度及多种结构参数估算出酵母菌3号染色体上78对位点之间的空间距离,进而首次建立起基于3C数据的染色体空间构象三维模型。2011年,Rousseau等[66]提出一种适用于5C和Hi-C数据的染色体空间结构三维建模方法MCMC5C,该方法给出从染色体交互频率到位点空间距离的概率模型,采用马尔可夫链蒙特卡罗抽样算法进行求解,并将其用在1Mb分辨率的Hi-C数据集上,建立起人类14号染色体的三维模型。2013年,Zhang等[67]提出基于Hi-C数据的染色体三维结构建模方法ChromSDE,其借助黄金分割搜索算法对交互频率与空间距离之间的转换进行参数优化,利用半正定规划技术建立起染色体三维结构模型。2013年,华中农业大学彭城等[68]提出一种基于Hi-C数据的染色体三维结构建模方法AutoChrom3D,其借助测序序列偏置松弛结构参数和分段线性函数实现各位点空间距离的转换,建立起染色体的三维结构模型,不同于以往其它建模方法,该方法考虑了不同实验中测序深度所引发的区域交互作用的偏差。2015年,Trieu等[69]在已有单染色体三维结构建模方法的基础上,提出了基因组三维结构建模软件MOGEN,该软件能够有效处理噪声以及不同染色体间Hi-C数据的差异。2017年,Paulsen等[70]提出了基于Hi-C数据和核纤层蛋白Chip-seq数据的全基因组三维结构建模软件Chrom3D,相比于之前的同类方法,Chrom3D集成了TADs径向位置约束条件,具备在单细胞水平进行全基因组三维空间结构建模的能力。2018年,Segal等[71]针对现有染色体三维结构重建算法准确性难以评估的现状,提出了基于染色体结构图谱的新的精度评估方法。同年,清华大学曾坚阳团队[72]提出一种基于构象能和流行学习的染色体三维结构建模框架GEM,与其它同类建模方法相比,GEM综合考虑了Hi-C交互数据以及染色体的生物物理可行性和结构稳定性,在方法有效性和模型物理生物特性验证中具备优势。可以看出,上述染色体三维空间结构建模方法,大多基于两步走的思路,即首先由交互频率数据推算出染色体各位点之间的空间距离,然后借助空间距离数据构建出染色体的空间结构模型,如MCMC5C、ChromSDE、AutoChrom3D和Chrom3D。与其形成对比的是不需要先行估算出各位点之间的空间距离,而是一种基于交互频率数据的结构模型优化过程,如MOGEN和GEM。
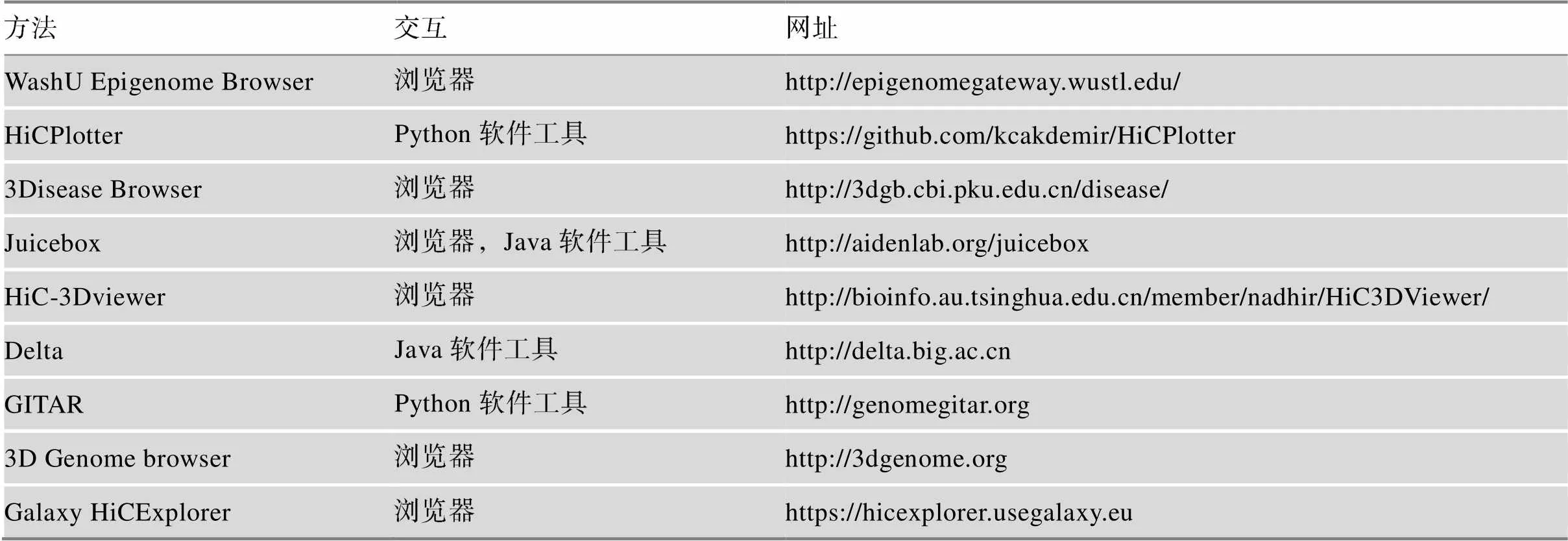
表4 Hi-C数据可视化软件
2 Hi-C研究发展动态
从2009年Hi-C技术的首次提出,到实验数据的分析处理,基于Hi-C技术的染色体空间结构研究历经了大约10年时间。10年来,Hi-C数据的生物信息学分析进展迅速。
在数据标准化方面,2012年到2013年期间,快速涌现出SCN[22]、ICE[24]和KR[25]等多种基于矩阵平衡策略的隐式标准化方法,以及以HiCNorm[23]为代表的基于模型构建策略的显式标准化方法。在随后的几年内,虽然也出现了多种接触矩阵标准化方法,如通过移除拷贝数偏差对ICE进行改进的caICB[26]方法,但这些方法均局限于对单样本Hi-C接触矩阵的处理。直到2018年,HiCcompare[27]方法首次将Hi-C接触矩阵标准化推向了双样本层面。2019年,其又被提出者升级为能够满足重复性样本标准化的MultiHiCcompare[28]方法。同年,出现了在配对末端序列水平上的标准化方法Binless[29]。可以看出,尽管各Hi-C数据标准化方法在显式和隐式,矩阵平衡和模型构建,以及接触矩阵水平和配对末端序列片段水平上有所差异,但基本呈现出由单样本标准化向跨样本标准化推进的发展动态。
在多级结构方面,2009年,Hi-C接触矩阵中的A/B区室被发现,首个A/B区室计算方法被提出[1]。同年,基于3C技术的染色质环结构被发现,其在基因表达调控中的作用被分析。2012年,接触矩阵中的TADs结构被发现,首个TADs边界点识别算法DI被提出[34]。2013年,TADs的层级结构被发现[35]。此后,涌现出多种Hi-C接触矩阵中各级结构的识别分析方法。如2014年的染色质环识别方法Fit-Hi-C[50]、HiCCUPS[51]和HIPPIE[52],2015年的多个细胞系下染色体A/B区室预测方法,同年的首个不同条件下染色质环差异分析方法dffHiC[53],2016年的首个层级式TADs识别方法TADtree[40],2017年的层级式TADs识别方法TADbit[41]、HiTAD[42]、IC-Finder[43]和GMAP[44],以及2018年首个支持TADs边界点差异性分析的方法HiCDB[47]。可以看出,接触矩阵中多级结构的探索大体经历了从结构发现到识别分析的过程。虽然A/B区室结构最早被发现,但对多级结构的分析更多集中在TADs和染色质环上,其中,对TADs的研究已从接触矩阵对角线上TADs边界点的识别,逐步深入到TADs层级式结构及其功能分析,而对染色质环的研究则呈现出由结构识别预测向不同条件下结构差异分析逐步推进的发展态势。
在数据可视化方面,自Hi-C技术提出之时就已使用log比例上的热图来显示接触矩阵数据。此后,在2013年,原Web Server服务器WashU Epigenome Browser[57]通过升级,具备了Hi-C数据热图显示及其功能可视化关联分析的功能。随着Hi-C数据的持续累积及其分析处理复杂度的不断提升,2016年,出现了Hi-C数据云平台及可视化分析软件Juicebox[60],以及集成Hi-C数据、Chip-seq数据和CR数据,且支持重排区域三维可视化的Web Server服务器3Disease Browser[59],为Hi-C相关数据的获取、结果关联分析以及三维可视化提供了软件工具支撑。2017年,出现了交互式三维立体可视化Web Server服务器HiC-3Dviewer[61],以及集成多种Hi-C数据分析工具的Web Server服务器Delta[62]。2018年,进一步涌现出集成多种Hi-C相关数据及其分析工具的可视化软件,如GITAR[63]、3D Genome browser[64]和Galaxy HiCExplorer[65]。可以看出,Hi-C数据可视化软件呈现出数据类型复杂化多样化、视觉交互三维立体化以及分析工具集成化的发展态势。
在三维建模方面,2011年,适用于5C和Hi-C数据的染色体空间结构三维建模方法MCMC5C[66]被提出。此后,专注于Hi-C数据的染色体三维建模方法陆续出现,如2013年的ChromSDE[67]和AutoChrom3D[68]。此几种方法虽然在考量因素和具体算法上有所不同,但均遵循了从交互频率到染色体各位点空间距离推算,再到染色体空间结构建模的同一思路。随后,出现了一类无需先行估算各位点空间距离,而是直接基于Hi-C交互频率数据进行染色体空间结构建模的方法,如2015年的全基因组三维结构建模方法MOGEN[69],以及2018年基于构象能和流行学习的建模方法GEM[71]。此外,2017年,出现了支持在单细胞水平上进行全基因组Hi-C三维建模的方法Chrom3D[70]。可以看出,基于Hi-C数据的染色体三维建模方法,经历了由分步计算到直接建模,由单个染色体向全基因组,再向单细胞水平逐步拓展的发展过程(图3)。
3 Hi-C研究发展趋势
从上述研究现状和发展动态可以看出,Hi-C技术及实验数据分析已经成为三维基因组学中备受关注的问题。以下仅从4个方面对Hi-C生物信息学方向的研究趋势进行浅析,包括跨样本标准化、多级结构差异及其调控机制分析、单细胞Hi-C数据分析和Hi-C数据可视化平台。
3.1 跨样本标准化
Hi-C数据的标准化绝大多数是在接触矩阵水平上展开。接触矩阵上的标准化方法按照是否考虑系统偏差的具体来源类型可分为显式和隐式标准化,按照各样本间是否存在数据交互又可分为单样本和跨样本标准化。标准化是后续分析的数据质量保障,而单样本标准化方法无法保障两组重复性样本之间的统计可比性,仅有的跨样本标准化方法HiCcompare和MultiHiCcompare,在接触矩阵分辨率不断提高、重复性样本数量持续增加和后续分析处理日趋复杂化的情况下,面临质量、效率和方法选择上的多重压力。因此,适用于高分辨率的跨样本高效标准化方法的研究,是Hi-C数据后续分析的结果质量保证和必经之路。
3.2 多级结构差异及其调控机制分析
差异分析是研究基因表达调控的重要手段之一,其通过分析不同条件下两组样本之间的显著性差异,探索基因和表型之间的联系。在基于Hi-C的三维基因组学中,染色体的多级结构与基因表达调控息息相关,使得不同条件下多级结构的差异分析成为此新领域的核心问题之一。如上述Hi-C研究现状和发展动态所述,目前,虽然已涌现出多种用于A/B区室、TADs和染色质环分析的方法软件,但具有差异分析能力的方法却十分缺乏,如可用于Hi-C接触矩阵中染色质环差异分析的方法diffHic。大多差异分析仍停留在单样本实验验证探索或者简单统计分析阶段。例如,Fraser等[73]在2015年给出小鼠胚胎干细胞分化过程中不同时间点上层级式TADs的树形结构,并采用协表相关系数,分析了不同细胞系下TADs树形结构的构造差异。随着接触矩阵分辨率的不断提高,各级结构的可预测数目迅猛增长,再加上为降低随机误差而引入的重复性样本,单靠热图对比和统计检验已远远不能满足后续差异分析的需要,因此,在三维基因组学,研究不同条件下,包括两种正常细胞系之间、正常细胞系与癌变细胞系之间以及同一细胞系不同时间点之间,Hi-C数据中多级结构,包括A/B区室、TADs和染色质环,的差异分析方法,进而探索各级差异性结构在基因表达调控中作用机制,是探索生物体细胞分化、形态产生和疾病发生发展等不可或缺的手段,其必将成为未来Hi-C领域生物信息学的研究热点之一。

图3 Hi-C研究发展动态
3.3 单细胞Hi-C数据分析
单细胞Hi-C技术用于稀少细胞或者处于特殊形态细胞的染色体构象捕获。常规Hi-C技术只能借助群体细胞构象数据的平均值来估计染色体交互作用,个别细胞的重要信号往往会被当作异常值受到弱化,而单细胞Hi-C技术可以很好解决细胞群体的异质性问题,其通过对生命活动最小单位的空间构象进行捕获,得到更有针对性的染色体交互信息。自Takashi等[10]于2013年提出单细胞Hi-C技术以来,单细胞Hi-C数据分析也应运而生。例如,Liu等[74]于2018年提出用于消除单细胞Hi-C数据中系统性偏差的软件包scHiCNorm;Liu等[75]于2019年利用单细胞测序揭示了与骨髓基质细胞亚群和培养时间相关的基因表达特征。单细胞Hi-C技术使得在单细胞水平进行染色体空间交互作用的研究成为可能,极大推进了三维基因组学的发展,基于该项技术的单细胞Hi-C数据分析,使得不同条件下各类细胞之间的空间构象得以精细区分,对探究基因表达调控的时空机制意义重大,势必受到专家学者的广泛关注与重视。
3.4 Hi-C数据可视化平台
随着Hi-C数据中各级结构及其生物学功能分析的不断深入,可视化平台也面临诸多挑战,逐步朝着数据复杂化多样化、视觉交互三维立体化以及分析工具集成化方向发展。Hi-C数据的复杂化多样化,即各物种不同组织和不同细胞系下Hi-C数据和各类组学数据的整合、关联与显示,包括不同分辨率Hi-C数据和ChIP-seq、RNA-seq、SNP以及疾病相关CR等数据;视觉交互三维立体化,即交互作用数据的可视化已不再局限于传统热图形式,呈现出与三维建模相结合的交互式三维立体显示趋势;分析工具集成化,即各类用于Hi-C数据分析的基础性方法工具逐渐被集成到系统平台当中,如标准化方法以及A/B区室、TADs和染色质环预测方法等。此外,Hi-C数据的集约型分析显示方法也日趋重要。得益于Hi-C技术的进步,接触矩阵的分辨率得到了显著提高,从原来的Mb级别发展到现今的1 Kb甚至200 bp[76],这使得高分辨率条件下Hi-C数据的处理显示面临计算资源不足的压力,因此,高效快速的Hi-C数据组织、分析及可视化方法工具在平台集成中将更具优势。
[1] Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long- range interactions reveals folding principles of the human genome., 2009, 326(5950): 289–293.
[2] Schmitt AD, Hu M, Ren B. Genome-wide mapping and analysis of chromosome architecture., 2016, 17(12): 743–755.
[3] Li GL, Ruan YJ, Gu RS, Du SM. Emergence of 3D genomics., 2014, 59(13):1165–1172.李国亮, 阮一骏, 谷瑞升, 杜生明. 起航三维基因组学研究. 科学通报, 2014, 59(13): 1165–1172.
[4] Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation., 2002, 295(5558): 1306–1311.
[5] Zhao ZH, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions., 2006, 38(11): 1341–1347.
[6] Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements., 2006, 16(10): 1299–1309.
[7] Zhang XY, He C, Ye BY, Xie DJ, Shi ML, Zhang Y, Shen WL, Li P, Zhao ZH. Optimization and quality control of genome-wide Hi-C library preparation., 2017, 39(9): 847–855.张香媛, 何超, 叶丙雨, 谢德健, 师明磊, 张彦, 沈文龙, 李平, 赵志虎. 全基因组染色质相互作用Hi-C文库制备的优化及其质量控制. 遗传, 2017, 39(9): 847–855.
[8] de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization., 2012, 26(1): 11–24.
[9] Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares- Cadete F, Sugar R, Javierre BM, Nagano T, Katsman Y, Sakthidevi M, Wingett SW, Dimitrova E, Dimond A, Edelman LB, Elderkin S, Tabbada K, Darbo E, Andrews S, Herman B, Higgs A, LeProust E, Osborne CS, Mitchell JA, Luscombe NM, Fraser P. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements., 2015, 25(4): 582–597.
[10] Takashi Nagano, Yaniv Lubling, Tim J. Stevens, Stefan Schoenfelder, Eitan Yaffe, Wendy Dean, Ernest D. Laue, Amos Tanay, Peter Fraser. Single-cell Hi-C reveals cell-to- cell variability in chromosome structure., 2013, 502(7469): 59–64.
[11] Liang ZY, Li GP, Wang ZJ, Djekidel MN, Li YJ, Qian MP, Zhang MQ, Chen Y. BL-Hi-C is an efficient and sensitive approach for capturing structural and regulatory chromatin interactions., 2017, 8(1): 1622.
[12] Lin D, Hong P, Zhang SH, Xu WZ, Jamal M, Yan KJ, Lei YY, Li L, Ruan YJ, Fu Z, Li GL, Cao G. Digestion- ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture., 2018, 50(5): 754–763.
[13] Barrett T, Edgar R. Gene expression omnibus: microarray data storage, submission, retrieval, and analysis., 2006, 411: 352–369.
[14] Qu HZ, Fang XD. A brief review on the human encyclopedia of DNA elements (encode) project., 2013, 11(3): 135–141.
[15] Moore D, Dines J, Doss MM, Vepa J, Cheng O, Hain T. Juicer: A weighted finite-state transducer speech decoder., 2006, 4299: 285–296.
[16] de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization., 2012, 26(1): 11–24.
[17] Shavit Y, Merelli I, Milanesi L, Lio’ P. How computer science can help in understanding the 3D genome architecture., 2016, 17(5): 733–744.
[18] Schmitt AD, Hu M, Ren B. Genome-wide mapping and analysis of chromosome architecture., 2016, 17(12): 743–755.
[19] Eagen KP. Principles of chromosome architecture revealed by Hi-C., 2018, 43(6): 469–478.
[20] Zhang XL, Fang H, Wang XW. The progress of methods for analysing 3D genome data., 2018, 45(11): 1093–1105.张祥林, 方欢, 汪小我. 三维基因组数据分析方法进展. 生物化学与生物物理进展, 2018, 45(11): 1093–1105.
[21] Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture., 2011, 43(11): 1059–1065.
[22] Cournac A, Marie-Nelly H, Marbouty M, Koszul R, Mozziconacci J. Normalization of a chromosomal contact map., 2012, 13(1): 436.
[23] Hu M, Deng K, Selvaraj S, Qin ZH, Ren B, Liu JS. HiCNorm: removing biases in Hi-C data via poisson regression., 2012, 28(23): 3131–3133.
[24] Imakaev M, Fudenberg F, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA. Iterative correction of Hi-C data reveals hallmarks of chromosome organization., 2012, 9(10): 999–1003.
[25] Knight PA, Ruiz D. A fast algorithm for matrix balancing., 2013, 33(3): 1029–1047.
[26] Wu HJ, Michor F. A computational strategy to adjust for copy number in tumor Hi-C data., 2016, 32(24): 3695–3701.
[27] Stansfield JC, Cresswell KG, Vladimirov VI, Dozmorov MG. HiCcompare: an R-package for joint normalization and comparison of Hi-C datasets., 2018, 19(1): 279.
[28] Stansfield JC, Cresswell KG, Dozmorov MG. multiHiCcompare: joint normalization and comparative analysis of complex Hi-C experiments., 2019, 35(17): 2916–2923.
[29] Spill YG, Castillo D, Vidal E, Marti-Renom MA. Binless normalization of Hi-C data provides significant interaction and difference detection independent of resolution., 2019, 10(1): 1938.
[30] Ning CY, He MN, Tang QZ, Zhu Q, Li MZ, Li DY. Advances in mammalian three-dimensional genome by using Hi-C technology approach., 2019, 41(3): 215–233.宁椿游, 何梦楠, 唐茜子, 朱庆, 李明洲, 李地艳. 基于Hi-C技术哺乳动物三维基因组研究进展. 遗传, 2019, 41(3): 215–233.
[31] Fortin JP, Hansen KD. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data., 2015, 16(1): 180.
[32] Dong PF, Tu XY, Chu PY, Lu P, Zhu N, Grierson D, Du BJ, Li PH, Zhong SL. 3D chromatin architecture of large plant genomes determined by local A/B compartments., 2017, 10(12): 1497–1509.
[33] Miura H, Poonperm R, Takahashi S, Hiratani I. Practical analysis of Hi-C data: generating A/B compartment profiles., 2018: 221–245.
[34] Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions., 2012, 485(7398): 376–380.
[35] Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong CT, Hookway TA, Guo CY, Sun YH, Bland NJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG. Architectural protein subclasses shape 3D organization of genomes during lineage commitment., 2013, 153(6): 1281–1295.
[36] Pope BD, Ryba T, Dileep V, Yue F, Wu WS, Denas O, Vera DL, Wang YL, Hansen RS, Canfield TK, Thurman RE, Cheng Y, Gülsoy G, Dennis JH, Snyder MP, Stamatoyannopoulos JA, Taylor J, Hardison RC, Kahveci T, Ren B, Gilbert DM. Topologically associating domains are stable units of replication-timing regulation., 2014, 515(7527): 402–405.
[37] Narendra V, Bulajić M, Dekker J, Mazzoni EO, Reinberg D. Corrigendum: CTCF-mediated topological boundaries during development foster appropriate gene regulation., 2016, 30(24): 2657–2662.
[38] Lévy-Leduc C, Delattre M, Mary-Huard T, Robin S. Two- dimensional segmentation for analyzing Hi-C data., 2014, 30(17): i386–i392.
[39] Shin HJ, Shi Y, Dai C, Tjong H, Gong K, Alber F, Zhou XJ. TopDom: an efficient and deterministic method for identifying topological domains in genomes., 2015, 44(7): e70.
[40] Weinreb C, Raphael BJ. Identification of hierarchical chromatin domains., 2016, 32(11): 1601– 1609.
[41] Serra F, Baù D, Goodstadt M, Castillo D, Filion GJ, Marti-Renom MA. Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors., 2017, 13(7): e1005665.
[42] Wang XT, Cui W, Peng C. HiTAD: detecting the structural and functional hierarchies of topologically associating domains from chromatin interactions., 2017, 45(19): e163.
[43] Haddad N, Vaillant C, Jost D. IC-Finder: inferring robustly the hierarchical organization of chromatin folding., 2017, 45(10): e81.
[44] Yu WB, He B, Tan K. Identifying topologically associating domains and subdomains by gaussian mixture model and proportion test., 2017, 8(1): 535.
[45] Norton HK, Emerson DJ, Huang H, Kim J, Titus KR, Gu S, Bassett DS, Phillips-Cremins JE. Detecting hierarchical genome folding with network modularity., 2018, 15(2): 119–122.
[46] Li AS, Yin XC, Xu BX, Wang DY, Han JM, Wei Y, Deng Y, Xiong Y, Zhang ZH. Decoding topologically associating domains with ultra-low resolution Hi-C data by graph structural entropy., 2018, 9(1): 3265.
[47] Chen FL, Li GP, Zhang MQ, Chen Y. HiCDB: a sensitive and robust method for detecting contact domain boundaries., 2018, 46(21): 11239–11250.
[48] Sexton T, Bantignies F, Cavalli G. Genomic interactions: chromatin loops and gene meeting points in transcriptional regulation., 2009, 20(7): 849–855.
[49] Lu YL, Zhou YP, Tian WD. Combining Hi-C data with phylogenetic correlation to predict the target genes of distal regulatory elements in human genome., 2013, 41(22): 10391–10402.
[50] Ay F, Bailey TL, Noble WS. Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts., 2014, 24(6): 999–1011.
[51] Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping., 2014, 159(7): 1665–1680.
[52] Hwang YC, Lin CF, Valladares O, Malamon J, Kuksa PP, Zheng Q, Gregory BD, Wang LS. HIPPIE: a high-throughput identification pipeline for promoter interacting enhancer elements., 2014, 31(8): 1290–1292.
[53] Lun ATL, Smyth GK. diffHic: a bioconductor package to detect differential genomic interactions in Hi-C data., 2015, 16(1): 258.
[54] Zhang H, Li FF, Jia Y, Xu BX, Zhang YQ, Li XL, Zhang ZH. Characteristic arrangement of nucleosomes is predictive of chromatin interactions at kilobase resolution., 2017, 45(22): 12739–12751.
[55] Djekidel MN, Chen Y, Zhang MQ. FIND: difFerential chromatin interactions detection using a spatial poisson process., 2018, 28(3): 412–422.
[56] Manduchi E, Chesi A, Hall MA, Grant SFA, Moore JH. Leveraging putative enhancer-promoter interactions to investigate two-way epistasis in type 2 diabetes GWAS., 2018, 23: 548–558.
[57] Zhou X, Lowdon RF, Li DF, Lawson HA, Madden PAF, Costello JF, Wang T. Exploring long-range genome interactions using the WashU epigenome browser., 2013, 10(5): 375–376.
[58] Akdemir KC, Chin L. HiCPlotter integrates genomic data with interaction matrices., 2015, 16(1): 198.
[59] Li RF, Liu YY, Li TT, Li C. 3Disease Browser: a web server for integrating 3D genome and disease-associated chromosome rearrangement data., 2016, 6: 34651.
[60] Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom., 2016, 3(1): 99–101.
[61] Djekidel MN, Wang MJ, Zhang MQ, Gao JT. HiC- 3DViewer: a new tool to visualize Hi-C data in 3D space., 2017, 5(2): 183–190.
[62] Tang BX, Li FF, Li J, Zhao WM, Zhang ZH. Delta: a new web-based 3D genome visualization and analysis platform., 2017, 34(8): 1409–1410.
[63] Calandrelli R, Wu QY, Guan JH, Zhong S. GITAR: an open source tool for analysis and visualization of Hi-C data., 2018, 16(5): 365–372.
[64] Wang YL, Song F, Zhang B, Zhang LJ, Xu J, Kuang D, Li DF, Choudhary MNK, Li Y, Hu M, Hardison R, Wang T, Yue F. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions., 2018, 19(1): 151.
[65] Wolff J, Bhardwaj V, Nothjunge S, Richard G, Renschler G, Gilsbach R, Manke T, Backofen R, Ramírez F, Grüning BA. Galaxy HiCExplorer: a web server for reproducible Hi-C data analysis, quality control and visualization., 2018, 46(W1): W11–W16.
[66] Rousseau M, Fraser J, Ferraiuolo MA, Dostie J, Blanchette M. Three-dimensional modeling of chromatin structure from interaction frequency data using markov chain monte carlo sampling., 2011, 12(1): 414.
[67] Zhang ZZ, Li GL, Toh KC, Sung WK. Inference of spatial organizations of chromosomes using semi-definite embedding approach and Hi-C data., 2013: 317–332.
[68] Peng C, Fu LY, Dong PF, Deng ZL, Li JX, Wang XT, Zhang HY. The sequencing bias relaxed characteristics of Hi-C derived data and implications for chromatin 3D modeling., 2013, 41(19): e183.
[69] Trieu T, Cheng JL. MOGEN: a tool for reconstructing 3D models of genomes from chromosomal conformation capturing data., 2015, 32(9): 1286–1292.
[70] Paulsen J, Sekelja M, Oldenburg AR, Barateau A, Briand N, Delbarre E, Shah A, Sørensen AL, Vigouroux C, Buendia B, Collas P. Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts., 2017, 18(1): 21.
[71] Segal MR, Bengtsson HL. Improved accuracy assessment for 3D genome reconstructions., 2018, 19(1): 196.
[72] Zhu GX, Deng WX, Hu HL, Ma R, Zhang S, Yang JL, Peng J, Kaplan T, Zeng JY. Reconstructing spatial organizations of chromosomes through manifold learning., 2018, 46(8): e50.
[73] Fraser J, Ferrai C, Chiariello AM, Schueler M, Rito T, Laudanno G, Barbieri M, Moore BL, Kraemer DCA, Aitken S, Xie SQ, Morris KJ, Itoh M, Kawaji H, Jaeger I, Hayashizaki Y, Carninci P, Forrest ARR, Semple CA, Dostie J, Pombo A, Nicodemi N. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation., 2015, 11(12): 852.
[74] Liu T, Wang Z. scHiCNorm: a software package to eliminate systematic biases in single-cell Hi-C data., 2017, 34(6): 1046–1047.
[75] Liu ST, Stroncek DF, Zhao YD, Chen V, Shi RY, Chen JG, Ren JQ, Liu H, Bae HJ, Highfill SL, Jin P. Single cell sequencing reveals gene expression signatures associated with bone marrow stromal cell subpopulations and time in culture., 2019, 17(1): 23.
[76] Wang Q, Sun Q, Czajkowsky DM, Shao ZF. Sub-kb Hi-C inreveals conserved characteristics of TADs between insect and mammalian cells., 2018, 9(1): 188.
Current status and future perspectives in bioinformatical analysis of Hi-C data
Hongqiang Lyu, Lele Hao, Erhu Liu, Zhifang Wu, Jiuqiang Han, Yuan Liu
The spatial interaction of chromosomes is regarded as an important issue affecting the regulation of gene expression, and the high-throughput chromosome conformation capture (Hi-C) technology has become the primary tool to explore the temporal and spatial interactions of chromosomes in three-dimensional genomics. With the continuous accumulation of Hi-C samples and the increasing complexity of pipelines, the bioinformatic analysis of Hi-C data has been considered an opportunity and a challenge for understanding the spatial regulation mechanism of gene expression. In this paper, the current status and development outline of bioinformatic methods for Hi-C data are introduced, including data normalization, multi-level structure analysis, data visualization and 3D modeling, especially of multi-level structure at A/B compartments, topological associated domains (TADs) and chromain looping levels. Based on this, we provide the outlook of future hotspots and trends in this area. Hopefully our insight will be beneficial for the exploration of gene expression regulation from the traditional linear model to the 3D mode.
3D genomics; Hi-C; bioinformatics
2019-07-23;
2019-11-26
国家自然科学基金青年科学基金项目(编号:61602367)资助[Supported by the National Natural Science Foundation of China (No. 61602367)]
吕红强,博士,副教授,研究方向:生物大数据分析与处理。E-mail: hongqianglv@mail.xjtu.edu.cn
10.16288/j.yczz.19-163
2019/11/28 9:13:59
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20191127.1304.006.html
(责任编委: 方向东)
