植物三维染色质构型研究进展
2020-03-04董芊里王金宾李晓宠宫磊
董芊里,王金宾,李晓宠,宫磊
综述
植物三维染色质构型研究进展
董芊里,王金宾,李晓宠,宫磊
东北师范大学生命科学学院,分子表观遗传学教育部重点实验室,长春 130024
染色质在细胞核内的缠绕、折叠及其在细胞核内的空间排布是真核生物染色质构型的主要特征。在经典DNA探针荧光原位杂交显微观察的基础上,基于新一代测序技术的Hi-C及ChIA-PET染色质构型捕获技术已经被广泛应用于动物及植物细胞核染色质构型的研究中,并以新的角度定义了包括:染色体(质)域(chromosome territory)、A/B染色质区室(compartment A/B)、拓扑偶联结构域(topological associated domains, TADs)、染色质环(chromatin loops)等在内的多个更为精细的染色质构型。利用以上两种主流技术,越来越多的植物物种染色质构型特征被鉴定、分析和比较。本文系统分析和总结了近年来以植物细胞为模型的细胞核染色构型领域取得的重要成果,包括各级染色质构型特征的组成、建立机制和主要影响因素等。在此基础上,分析了目前研究植物染色质构型技术的瓶颈和突破性的技术进展,并对后续研究主要关注的问题和研究内容进行了展望,以期为相关领域的研究提供更多的理论参考和依据。
染色质构型;高通量染色质构象捕获测序;末端配对标签测序;染色质区室;拓扑偶联结构域
染色质(chromatin)是由基因组DNA与组蛋白等结构性蛋白通过不同层级的缠绕而形成[1]。作为真核生物遗传物质的载体,染色质需要填充在微米级的狭小细胞核中。其缠绕、折叠及染色质在细胞核空间内的不同排布模式,组成了染色质空间构型(chromatin architectures)研究的主要内容。基于光学显微镜观察的DNA探针荧光原位杂交(fluorescencehybridization, FISH),是研究染色质构型的经典研究方法,该方法能够实现通过肉眼观察细胞内整体或特定区段的染色质空间构型[2,3]。但受限于技术本身的局限(DNA探针杂交的灵敏度等因素),FISH技术能够检测到的染色质构型的分辨率有限,且其不能量化染色质区段的空间距离。受益于近年来新一代测序和显微成像技术的发展,通过体外测序和体内原位观察的方法实现了染色质构型的精细定量和分析,也使得基于生物信息学的染色质构型研究成为目前的热点研究领域之一[4~7]。
目前,染色质构型研究的相关研究成果还主要集中在哺乳动物细胞系或其他动物模型中[8],鉴于植物和动物在细胞核结构和基因组组成等方面的差异,植物细胞中的染色质构型的模式、特征(不同植物组织和不同类型细胞中的保守性和特异性)以及形成机制等还亟需系统探讨[9,10]。本文在简要回顾近年来染色质构型研究技术发展的基础上,系统分析和总结了以植物细胞为模型的细胞核染色质构型领域取得的重要成果,阐述了植物染色质构型研究领域内目前的技术瓶颈,并探讨了后续研究中主要关注的问题和发展前景,以期为相关领域的研究提供更多的理论参考和依据。
1 染色质构型研究技术
目前,三维基因组研究领域内最为主流的染色质构型研究技术,包括:断层扫描电镜成像(chromosome electron microscopy tomography)、高通量染色质构象捕获(high-throughput chromosome conformation capture, Hi-C)以及末端配对标签测序(paired- end tag sequencing, ChIA-PET)[11,12]。其中,电镜成像虽然可以在极高的分辨率下直接解析染色质结构,但由于其检测到的染色质互作区段有限,因此本文未将其列为介绍的内容,主要对Hi-C和ChIA-PET两种染色质构型分析技术进行详细介绍。
1.1 Hi-C技术原理
2002年,Dekker等[13]开发了染色质构象捕获技术(chromosome conformation capture, 3C)。3C技术首先是用甲醛瞬时固定细胞核染色质,用限制性内切酶酶切消化染色质—蛋白质交联物。随后,在DNA浓度极低的条件下,使用高浓度连接酶连接消化物,使得交联在一起的DNA片段粘性末端在交联片段之间重新连接。最后,使用蛋白酶消化交联物进而释放出结合的蛋白质,并根据实验目的的不同,用推测可能有互作的目的片段的引物进行普通PCR或定量PCR扩增,最终确定其是否存在显著的相互作用。该技术是一种“一对一”(OneOne)的检测染色质相互作用的技术,即对于基因组上两个已知的目的染色质区段,利用3C技术可以得知这两个片段的相互作用情况。以3C技术为基础,2009年,Lieberman-Aiden等[11]将3C技术与新一代测序技术(next-generation sequencing, NGS)技术结合,研发出了高通量染色质构象捕获方法,即Hi-C (high- throughput chromosome conformation capture)技术(图1A)。虽然Hi-C技术脱胎于3C技术,但整个操作过程相对于3C技术更加的冗长和繁琐,针对此类问题,赵志虎等对Hi-C实验流程中的关键步骤进行了优化和改进,促进了Hi-C技术进一步的广泛应用[14]。Hi-C技术分析步骤主要包括:(1)甲醛交联:使用甲醛瞬间固定细胞核染色质。使得细胞内染色质交联,生成DNA-蛋白质复合物或蛋白质–蛋白质复合物;(2)酶切:使用限制性内切酶消化染色质,通常使用六碱基内切酶(6-cutter),如d Ⅲ、Ⅱ、或R Ⅰ等,或四碱基内切酶(4-cutter),如Ⅰ、Ⅱ或Ⅰ等;(3)生物素标记:用生物素标记的核苷酸片段,连接至酶切时产生的粘性末端;(4)片段末端链接:利用DNA连接酶,将不同的切口末端连接起来,形成环状嵌合分子;(5)剪切并纯化:利用机械力打断环状嵌合分子,产生小片段DNA。并利用亲和素特异性结合生物素的原理,将携带生物素核苷酸的DNA片段富集;(6)文库构建及NGS测序:将携带生物素核苷酸的DNA片段两端添加测序接头,构建Hi-C测序文库,利用Illumina NGS测序平台进行高通量测序,并将测序结果转化为相应大小的染色质区段之间的相互作用矩阵和热图。
1.2 ChIA-PET技术原理
2009年,Fullwood等[12]开发了染色质远程交互测序技术(chromatin interaction analysis by paired- end tag sequencing, ChIA-PET)。ChIA-PET是基于免疫共沉淀(chromatin immunoprecipitation, ChIP)、染色质邻近连接(chromatin proximity ligation)、配对末端标签(paired-end tags)以及新一代测序技术,所开发出的一种分析全基因组范围内远程染色质相互作用的新技术。与Hi-C技术相比,ChIA-PET在甲醛交联固定后,优先进行ChIP富集;随后的操作中不同于Hi-C实验中使用的限制性内切酶消化,ChIA- PET则使用超声破碎进行物理打断。后续的操作步骤基本与Hi-C技术相似,即连接生物素并连接、纯化以及建库并测序(图1B)。

图1 Hi-C及ChIA-PET技术流程示意图
A:Hi-C技术流程示意图;B:ChIA-PET技术流程示意图。
1.3 Hi-C与ChIA-PET技术的特征和应用
鉴于Hi-C技术与ChIA-PET技术在原理层面既存在相似又有不同,因此两种方法有着各自的优点与局限。有关两种技术的特征和应用范围的具体比较,详见表1。
2 植物不同等级的染色质构型特征
作为基于概率模型分析染色质构型的主流技术,Hi-C和ChIA-PET已经被广泛应用在不同植物的染色质构型特征的鉴定、功能分析以及相互关系的研究中(表2)。按照目前业内广泛接受的标准(包含染色质区段大小的尺度和功能特征),植物染色质构型涵盖的等级主要包括:染色体(质)域(chromosome territory, CT)、A/B染色质区室(compartment A/B)、拓扑偶联结构域(topological associated domains, TADs)、染色质环(chromatin loops)以及IHIs/ KEEs (interactive heterochromatic islands/KNOT ENGAGED ELEMENTs)[5,16](图2)。接下来将从植物物种、材料性质、研究方法(Hi-C或ChIA-PET)、组成、建立机制或者主要的影响因素等方面,详细说明植物不同等级染色质构型的主要特征。

表1 Hi-C与ChIA-PET技术的特征和应用范围的比较

表2 植物不同等级的染色质构型特征汇总
2.1 染色体(质)域CT
在细胞分裂间期(多数细胞主要处于的时期),虽然植物染色质主要呈现较为松散的状态,但在整个细胞核空间内仍呈现非随机的排布方式。每条染色质占有自己特有的一个细胞核空间CT。CT的概念,最早是由Rabl在1885年观察火蜥蜴(Salamander)细胞分裂的过程中提出;而后在1909年,由Boveri[31]对CT概念进行了进一步的凝练;在20世纪80年代,在人的细胞中利用染色体特异性的DNA探针完成的FISH观察,最终确认了CT的存在[1,32]。而植物CT的存在,首先是在拟南芥()中,利用细菌人工染色体(bacterial artificial chromosome)片段标记探针后,由FISH观察而确认的[1,2]。利用相似的FISH观察方法,通过特异性标记着丝粒、端粒以及其他染色体区段的BAC文库探针,不同植物物种中的CT排布特征被详细分析和归纳为不同的染色质构象(图3,A和B分别展示的Rabl和Rosette构象)。
利用Hi-C数据结合后期3D模型重构的方法(chromatin 3D remodeling),2009年Lieberman-Aiden等[11]首次应用成功绘制出了1M分辨率下人类淋巴母细胞(GM06990)的三维染色体结构,并验证了之前通过FISH技术发现的CTs的存在。利用已有的高分辨率Hi-C数据,本文也重构了拟南芥混合细胞类型平均呈现的3D染色质构象(图4)。与经典的FISH研究结果一致[1,33,34],拟南芥间期细胞主要呈现(混合细胞平均呈现出的相似特征)莲座状构象(Rosette configuration):端粒在核仁区聚集,着丝粒围绕在外围(图3B,图4)。这一构象特征与Hi-C互作热图显示的互作状态一致。前期FISH研究发现,拥有较大基因组的物种,如:玉米()、大麦()等,其间期细胞的染色质主要呈现Rabl构象(Rabl configuration):端粒和着丝粒分处于细胞核的两级,不同染色质的着丝粒左右的染色质臂之间紧密贴合(图3A)。近期玉米叶肉细胞(单一细胞类型)的Hi-C互作热图显示,其不同染色质的着丝粒和端粒之间的确呈现高强度的互作,而且不同染色质的着丝粒左右的染色质臂之间也一致性的存在染色质内及染色质之间的紧密互作。

图2 植物中染色质层次结构示意图
单个染色体在核中占据染色体(质)域(chromosome territories)的一个子空间。染色体(质)域可以进一步划分为不同的A/B染色质区室(compartment A/B)。拓扑偶联结构域内(TADs)的基因组区域显示出更强的相互作用,而它们与拓扑偶联结构域之外的相邻区域相互作用则相当有限。TADs内调节元件与其靶基因座连接形成的染色质环[10]。
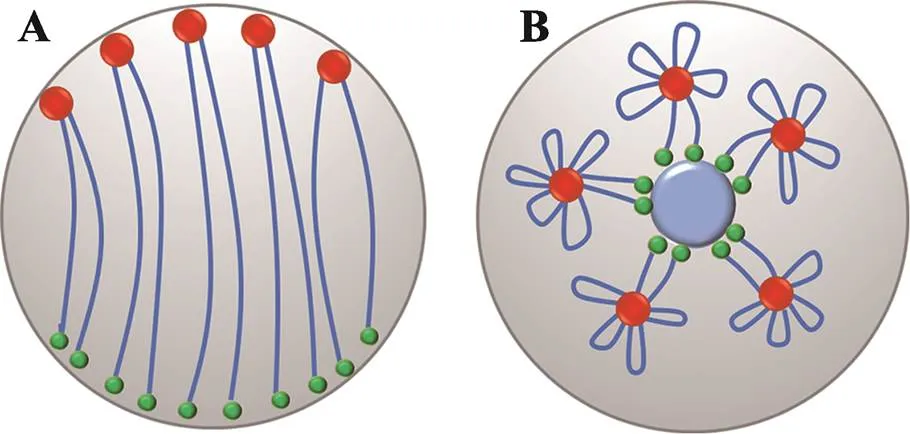
图3 Rabl及Rosette构象的构象展示
A:Rabl构象示意图;B:Rosette构象示意图。红色圆圈:近着丝粒区段;绿色圆圈:端粒区段;蓝色线段:染色体臂;蓝色圆圈:核仁。
然而,考虑到相同物种的不同细胞类型可能拥有不同的CT特征(呈现不同染色质构象),混合细胞类型的Hi-C测序数据不能够准确反映特定细胞呈现的CT染色质构象特征,故而后者需要更为精准的单细胞Hi-C技术(scHi-C)。近期,Zhou等[29]对水稻卵细胞、精子细胞、单细胞合子和芽叶肉细胞,进行了scHi-C测序和分析。通过3D模型重构,发现水稻卵子和精子细胞的染色质结构与叶肉细胞的染色质结构相当,并且在受精后得以重组。类似的scHi-C技术为在植物系统中更为精细的研究包括CT染色质构象在内的各层级染色质构型创造了条件。

图4 拟南芥Hi-C数据进行的染色体三维模拟结构图
基于Liu等[35]已发表的拟南芥Hi-C数据,进行染色体三维结构模拟。A:拟南芥5条染色体三维结构图;B:红色部分为着丝粒区域,绿色球体为端粒区域。可以看到基于Hi-C数据模拟出的染色体三维结构图与Rosette结构一致。
2.2 A/B染色质区室
A/B染色质区室概念,最早是由Lieberman- Aiden等[11]于2009年发表Hi-C技术时提出。其在研究中,发现同一条染色体内的所有染色质区段的互作热图总体呈现“格子(plaid)”式排布。在此观察结果的基础上,作者对同一染色体内所有染色质区段之间的Hi-C互作值,以染色质区段为单位计算其与其他区段之间互作的相关性,并利用主成分分析(principal component analysis,以下简称PCA)将具有相似互作模式的染色质区段(与同一染色体内的其他染色质区段的互作)划归成“正”和“负”两个特征向量值组(eigenvalues),各组内的染色质区段分别对应A和B两个染色质区室。相同类型区室内的染色质区段呈现更相似的染色质互作模式。除以上依据Hi-C数据定义A/B染色质区室之外,Wang等[36,37]使用单细胞水平的FISH技术,也在细胞内成功验证了经由Hi-C划分出的区室A和区室B在细胞核中存在空间上的分离。如图5所示,水稻日本晴Chr.2互作热图上可以明显观察到特征向量的正负交替,即A/B区室的交替分布。
近期的研究发现,植物物种的A/B染色质区室总体呈现与动物模型相似的染色质互作、基因组组成和表达以及表观遗传修饰的特征。首先,在染色质互作方面,植物物种中的同一条染色体内部,染色质区室B的不同染色质区段之间的互作用强度显著高于区室A的染色质区段[18,27,28,36]。但不同染色体间的染色质区室A的染色质区段之间的相互作用强度显著高于区室B的不同染色质区段之间的互作。以上互作状态表明,染色质区室A的染色质的折叠状态更加松散,并且区室A的染色质区段应主要排布在相应染色体CT的外围区域[27,36];其次,在基因组组成和表达方面,染色质区室A内的基因组片段更加显著富集基因,而染色质区室B内的基因组片段反而显著富集转座子(transposable elements, TEs)。相应的染色质区室A内更高密度的基因拥有更高的转录表达水平;最后,在表观遗传修饰模式和水平方面,激活性组蛋白修饰标记(如H3K4me2、H3K4me3和H3K9ac等)在染色质区室A中显著富集,而抑制性组蛋白修饰(如H3K9me2)和甲基化的胞嘧啶(CG、CHG和CHH三种序列环境)在区室B中显著富集。综上所述,在常染色质和异染色质区段划分较为明确的植物物种中(如水稻、西红柿、高粱、棉花等)[15,23,28],由于常染色质和异染色质往往分别富集在染色质区室A和B中,同一染色体内的染色质区室A和B之间频繁交替出现的情况较少(non-interlaced; interlace);相反的,在常染色质和异染色质区段没有明确划分的植物物种中(如拟南芥),同一染色体内的染色质区室A和B之间存在频繁的交替(interlaced)[29]。
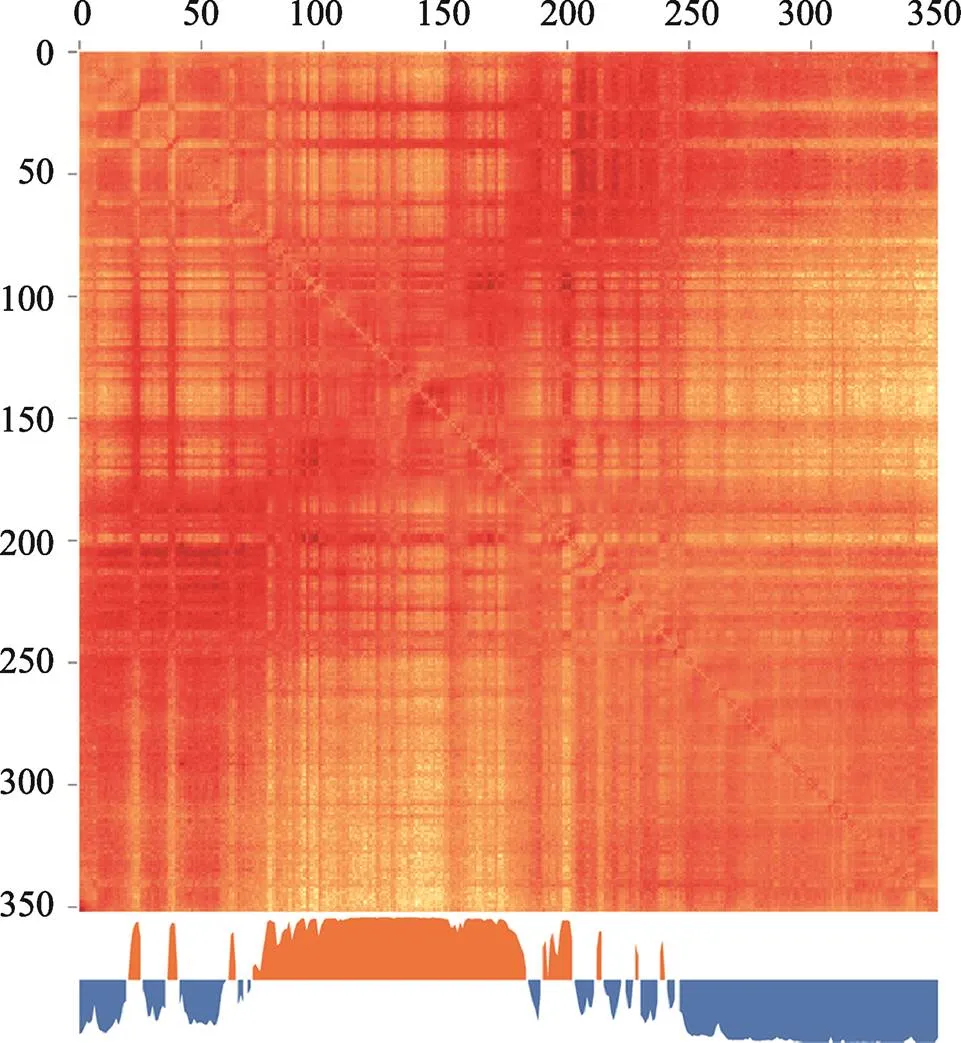
图5 水稻2号染色体(Chr.2)中A/B染色质区室的划分及其对应的互作热图
在其他以动物为模型的染色质构型研究中,不同细胞类型[38]、同一细胞类型的不同发育阶段(如受精卵的不同发育阶段)[39]以及不同处理条件下,同一染色质区段所在的染色质区室类型会发生A和B之间的转换[40]。对目前已经发表的植物染色质构型的研究结果进行总结分析后发现:(1)水稻混合细胞的Hi-C分析发现在冷胁迫处理后染色质区室A和B总体呈现较为稳定的状态[36];(2)水稻单细胞scHi-C分析发现卵细胞、精子和叶肉细胞的同一染色质区段所在的染色质区室类型有差别[29];(3)相较于常见的二倍体拟南芥细胞核染色质,同源染色体组加倍后形成的拟南芥四倍体中,出现了染色质区室A和B之间的转换以及伴随发生的组蛋白H3K4me3和H3K27me3修饰水平的变化[21];(4)对芸薹属()内代表性二倍体物种芜菁()和甘蓝()的染色质Hi-C构型数据分析发现,二者共线性区段内多数(61.62%)的染色质区室呈现保守状态(维持相同的染色质区室类型),但仍有约38.38%的区段发生了染色质区室类型的转换[22];(5)棉属()内自然进化过程中的异源多倍化(allopolyploidy)介导的不同亲本基因组融合和加倍,引发了其对应亲本亚基因组在多倍体陆地棉和海岛棉(和)中的染色质区段发生染色质区室A和B之间的转换[23]。
2.3 拓扑偶联结构域TADs
上述介绍的CTs和compartment A/B层级结构,均是在较大的分辨率上(>500 kb的染色质区段)定义和观察到的染色质构型。基于足够分辨率的Hi-C染色质互作数据,Dixon等[41,42]率先在人类和小鼠中定义并解析了更精细层级的一种染色质构型单元—TADs:同一TAD内的不同顺式染色质区段呈现更强的区域内互作,而这些区段与其他相邻染色质区段(不在同一TAD内)的互作较弱。据此,TADs是染色质上自身相互作用很强,而和临近的染色质区域相互作用受到抑制的结构单元;在Hi-C互作图谱热图(对称互作热图对角线下的一半热图)上,每个TAD对应相应染色质区段的互作呈现出颜色较深的三角形(图6A)。近期将超分辨率显微镜应用于动物单细胞染色质构型的研究也发现了类似TAD的染色质结构单元(TAD-like structural unit),该结构也呈现出与TAD类似的,结构单元内相互作用显著更强的染色质互作模式[43,44]。
目前,染色质TADs结构在动物细胞核内的形成机制[43~48]、组成和整体分布[42]、自身结构稳定性、维持基因组结构稳定性[49]、调控相应基因表达[42,50~53]等方面已经得到广泛研究和报道。对植物TADs在以上各个方面的特征总结发现:(1)哺乳动物染色质TADs主要是由CTCF(CTCCC-binding factor)和cohesin等绝缘蛋共同锚定于TAD边界处(TAD boundary),而TADs内部的cohesin来回滑动介导“loop extrusion”最终导致TADs的形成[54]。与果蝇()基因组相似,植物物种基因组虽然无编码CTCF的基因,但已发表的研究工作证实许多植物物种的染色质均可以形成TADs结构[15~17,19,20,22,24,25,34]。在目前已经鉴定的能够形成染色质TADs结构的植物物种中,包括水稻、玉米、番茄、高粱、棉花和甘蓝等,物种内能够参与形成TADs的保守性因子(类似CTCF的蛋白因子)还没有被鉴定出来。近期,Liu等[36]通过分析水稻TADs边界富集的DNA序列motif,获得了可能识别该DNA motif的蛋白因子—TCP转录因子(图6B),后者是否是形成TADs的候选保守因子,仍需后续的分子功能验证;(2)如上文提及,TADs的结构主要包含TAD边界和TAD内部(TAD-interior regions)两个区段。TADs内的两个结构区段所包含的DNA组成和其他表观遗传修饰的特征明显不同(植物和动物TADs一致):相较于TAD内部,TADs边界内的基因密度更高,染色质开放程度较高,转录表达水平较高;TAD边界和TAD内部往往分别富集激活(如H3K4me3等激活性组蛋白修饰等)和抑制性的表观遗传修饰(如H3K27me3等抑制性组蛋白修饰、5mC胞嘧啶甲基化等)[27,28,36]。此外,动物(以果蝇为例)和植物染色质TADs也呈现相似的在染色体上的分布状态,即激活性组蛋白修饰富集的转录激活TADs (active TADs)往往分布于常染色质区段;富集H3K9me2组蛋白修饰、HP1以及Su(var)3-9等蛋白的TADs (heterochromatin TADs)往往分布于异染色质区段;而富集H3K27me3组蛋白修饰和PcG (Polycomb group)蛋白的TADs (Polycomb- repressed TADs)除在异染色质区段内能检测到之外,也常在常染色区段出现[51];(3)在自身结构稳定性方面,动物TADs染色质结构在不同的细胞类型甚至于不同的物种之间都是相对保守的[41],但在生长发育过程中,同一细胞类型的TADs结构中的结构单元可能会出现合并(TADs merge)或者是分离(TADs split)的现象[39]。近期,植物中第一篇利用scHi-C技术探讨水稻单卵细胞、精子细胞、合子以及叶肉细胞染色质构型的研究,为分析同一植物物种的不同细胞类型和同一类型细胞的不同发育阶段、不同植物物种间的TADs染色质结构差异或动态变化,提供了实验技术及数据分析方面的强大支撑[29];(4)跨物种间的TADs位置的比较,能够反映出TADs维持基因组结构稳定的重要性。已有研究发现相对于人()的基因组组成,长臂猿()的物种形成和进化过程中会出现染色体的融合,而通过比较两个物种的Hi-C数据所鉴定出的TADs,结果发现TADs边界往往出现在长臂猿物种内的融合位点处。该结果暗示了长臂猿染色体结构的变异可能是由TADs边界区的染色质较为脆弱而造成或是由自然选择将携带有TAD内部染色体断裂或融合的个体筛选掉而造成[49]。目前,在植物中TADs是否及如何参与染色质结构变异的研究仅局限在多倍体棉花物种(陆地棉和海岛棉)以及芸薹属内二倍体芜菁和甘蓝的物种形成过程中。其中,Wang等[23]通过分析和比较多倍体陆地棉和海岛棉物种与现存的二倍体亲本的Hi-C染色质构型特征,发现多倍体化后的基因组其开放染色质区会偏倚性的出现TADs的重排或结构变化;Xie等[22]分析并比较了芜菁和甘蓝Hi-C数据能够检测到的TADs的组成,发现两个物种分化后,物种间的共线性区段内多数的TADs均呈现保守状态。除此之外,在其他植物物种分化过程中是否及如何发生TADs的组成变化及后者与基因组结构稳定性之间的关系却鲜有报道。这也将成为植物染色质构型研究领域内今后发展的一个重点研究方向;(5)在调控基因表达方面,从TADs染色质结构特征考虑,无论是在动物或者植物细胞核中,将细胞核内的染色质通过TADs结构的形式进行打包,可以摆脱线性距离较远的影响,从而拉近结构内两位点之间的空间距离,还可以隔绝结构单元内调控元件与外部靶点的相互作用,从而可以特异性识别结构内的靶点[41,42,55~58]。

图6 TAD结构及水稻TAD boundaries附近的互作和表观遗传修饰特征
A:TAD结构模式图;B:水稻TAD boundaries附近不同表达水平基因的分布以及各种组蛋白的修饰。
目前,在植物物种中TADs染色质结构作为一种重要且常见的染色质结构单元已经被广为接受,但是经典的模式植物拟南芥却是其中一个特例——其Hi-C染色质互作热图中没有经典的TAD互作模式[15~17,34]。有关拟南芥不存在经典TADs的理论猜测,主要来自于对其基因组组成的分析:模式植物拟南芥中的基因组相较于其他植物较小,其基因组内部含有较少的转座子(TEs)故而其染色体上的基因密度较高。据此,Liu等[5]推断,基因密度较低的植物物种,由于其基因在基因组上被转座子间隔开,因此需要形成局部的TADs染色质结构,才能够实现空间分离的基因的共表达调控。已经发现植物物种中的高转录密度和TADs结构的形成密切相关[5,36],而拟南芥的染色体臂上的转录密度则比较均匀,顺势调控区域往往位于启动子的附近,因此拟南芥中基因表达调控对TADs染色质结构的依赖性较弱。据此,以上有关基因组大小影响基因密度,进而影响染色质构型的推断得到了一定程度的支持。需要强调的是,以上提出的观点仅是基于已有研究的理论猜测,目前尚无定论。
2.4 染色质环
细胞核内比TADs更为精细的下一个层级的染色质构型特征是染色质环,这是一个更加局部的染色质结构单元。染色质环是一种非常常见的染色质内部的相互作用,该结构可以通过减小调控元件和靶基因之间的空间距离从而促进二者之间的相互作用[10,59]。在动物中的很多相关研究表明,主要发生在TADs结构内部的增强子和启动子之间的染色质环,在精确的转录调控过程中非常重要。动物中发现的一个增强子调控多个基因以及一个基因受到多个增强子的调控现象在植物物种鲜有报道[60]。
植物中不同的染色质环在不同的物种、同一物种的不同生长发育过程阶段,同样扮演着非常重要的角色[57,61~64]。通过分析单基因分辨率的拟南芥Hi-C数据,在常染色体臂上总共检测到了20 000多个染色质环[35],其中已知的少数染色质环在转录调控过程中发挥着重要作用[65~67]。而基于水稻高分辨率的Hi-C数据,研究人员共检测到了51 319个染色质环[27]。在这些染色质环结构中,转录起始位点(transcription start sites, TSSs)倾向与下游区域形成染色质环,而TTSs倾向与其上游形成环状结构[35]。这种现象类似于在酵母中报道的“gene globule”模型[68],并被后续定义为“基因自成环现象”。在拟南芥和水稻中的研究发现,自成环的基因往往要比未成环的基因更加活跃,且增强子–启动子形成的环状结构往往会激活其临近的基因[27]。
通过对5种植物基因组Hi-C的研究,科研人员发现玉米和高粱等具有较大基因组的物种拥有TADs结构外部的、长距离的染色质环状互作结构,其区别于动物TADs内较小的染色质环[28]。研究还发现,参与长距离染色质内部互作的区域往往和激活性的组蛋白修饰相关,并且富含基因。此外,对棉花的相关研究中,Wang等[23,24]发现,启动子区、潜在的增强子以及激活性表观遗传修饰的区域两两之间存在相互作用。他们将之分为启动子-启动子、增强子–增强子以及增强子-启动子等3种互作类型,并发现大部分参与的基因也都受到多个长距离相互作用的调节。最后,Lun等[15]利用高分辨率的ChIA-PET技术,捕获到了H3K4me3、RNAPII以及H3K9me2 3种靶蛋白的结合位点以及其介导的结合位点之间的相互作用,并将H3K4me3和RNAPII介导的染色质环分为PPI (promoter–promoter interaction) loop和BP (basal promoter) loop。该研究发现,PPI loop上的基因相较于BP loop上的基因表达量更高,且PPI loop上的基因在水稻基因组的进化过程中拥有更高的保守性;此外,H3K4me3和RNAPII共同介导的loop 基因相较于仅被K3K4me3或RNAPII介导的loop基因表达量更高;另外,H3K4me3以及RNAPII介导的染色质间的环状相互作用形成了复杂的空间转录单元,并且环状结构的形成促使了两端基因的共表达;最后,H3K9me2介导的染色质环(39~341 kb)相较于H3K4me3 (~18 kb)和RNAPII (11~27 kb)介导的染色质环更大。
2.5 IHIs/KEEs
利用Hi-C数据能够检测到植物细胞核特有的染色体内或染色体间的特殊染色质构型特征。在野生型拟南芥中,研究人员能够检测到10个非对角线的染色体(质)内或染色体(质)之间的强烈“网状”相互作用区段[18,19]。人们将这些强烈互作区域形成的结构称为KNOT,而将组成这种结构的区域命名为IHIs/KEEs[18]。对拟南芥中有关IHIs/KEEs的研究进行总结发现:(1)拟南芥的IHIs/KEEs是位于常染色体臂中的异染色质区域,富含TEs相关的重复序列、smRNAs以及H3K9me2、H3K27me1等异染色质标记[18,19];(2) Grob等[18]通过FISH技术证明KNOT结构在拟南芥细胞核中是真实存在的,并通过BLAT发现拟南芥中的IHIs/KEEs含有195 bp和70 bp的两个保守区域,前者对应于ATLANTYS3转座子(LTR/Gypsy家族),后者对应VANDAL6转座子(DNA MutR家族);(3)在拟南芥中IHIs/KEEs与端粒区存在着强烈的相互作用,而IHIs/KEEs与近着丝粒区虽然拥有着相似的序列特征和表观遗传修饰,但二者之间却并不存在显著的互作[18,19];(4)在拟南芥的表观遗传突变体中,IHIs/KEEs位点间的相互作用会发生动态变化[19]。在突变体中,IHIs/KEEs位点之间的相互作用增强。在和突变体中,IHIs/KEEs位点增多,但彼此之间的相互作用相对于野生型拟南芥来说出现了减弱的现象,在的“三突”拟南芥中,IHIs/KEEs位点同样出现了增多,但彼此之间的相互作用并没有发生显著的改变。以上结果表明,表观遗传修饰可能在IHIs/KEEs位点之间的相互作用中发挥着重要作用;(5) Grob等[18]猜测并证明了IHIs/KEEs位点是转座子转座插入的首选位点,该结果也暗示了IHIs/ KEEs在保护基因组的完整性中发挥着关键作用;(6)拟南芥中的端粒以及IHIs/KEEs并不会像其他异染色质区域会处在靠近细胞核膜的位置,而是会处在细胞核的内部[69]。
除以上在拟南芥中鉴定到的IHIs/KEEs位点,Dong等[27]通过Hi-C技术在水稻中也鉴定到了81个IHIs/KEEs位点,并发现最显著的IHIs/KEEs位点处在水稻的Chr.1和Chr.12。通过探究水稻中的IHIs/ KEEs位点的特征发现,水稻中的IHIs/KEEs与拟南芥相似,也同样富集TEs、sRNAs、H3K9me2以及DNA甲基化(尤其是CHH序列环境)。但与拟南芥中鉴定到的两个保守序列不同,水稻中的IHIs/KEEs位点只含有一个163 bp的保守序列。此外,水稻中的IHIs/KEEs富含多种转座子类型,包括ATLANTYS2、RIRE3、SZ-64B和TRUNCATOR2等,其中ATLANTYS2也存在于拟南芥的IHIs/KEEs位点中。
最近在芸薹属植物芜菁和甘蓝中,Xie等[22]利用Hi-C技术分别鉴定到了31个和7个IHIs/KEEs位点。其中,芜菁中的10条染色体都含有IHIs/KEEs位点分布。与拟南芥和水稻中鉴定到的IHIs/KEEs位点不同,芜菁中的IHIs/KEEs位点在一些染色体上是成簇存在的。因此,从基因组进化的角度看,芜菁中的IHIs/KEEs位点经历了扩张,而与之相反的是,在甘蓝中的IHIs/KEEs位点则出现了收缩减少的现象。
3 结语与展望
虽然目前三维基因组学技术的应用使植物染色质构型的研究取得了突飞猛进的进步,但仍存在一些瓶颈问题:(1)二倍体植物细胞核内的染色体是以同源染色体对的形式出现(即染色体核型组成为2n)。但目前所有已发表的利用Hi-C及ChIA-PET技术探讨染色体(质)构型的研究,均未能区分同源染色体对内的各单条染色体的染色质空间构型。例如,水稻细胞核内实际共含有24条染色体,目前得到的染色体互作结果是将两条同源染色体的互作混合的平均状态,并非细胞内完全真实的染色体互作情况;(2)在进行scHi-C建库时,由于DNA含量相对较少,无法获得更高分辨率的Hi-C互作热图。因此,在分析染色体(质)构型特征时,无法进行拓扑偶联结构域和染色质环水平的特征分析;(3)目前植物中拓扑偶联结构域部分的识别与鉴定主要依靠Dixon等[50]和Crane等[70]为动物拓扑偶联结构域开发的算法,或采用Liu等[36]在鉴定水稻拓扑偶联结构域时所使用的算法。业内仍亟需对以上算法在植物中应用的有效性、差异性和可适用范围进行评估,进而总结或开发出一个更适用于植物细胞核染色质拓扑偶联结构域鉴定的算法。
综上所述,染色质三维结构的特征及功能研究已经成为目前基因组学和细胞生物学研究的重要研究领域。随着鉴定和分析植物染色质构型技术的不断突破和进步(如scHi-C的出现),以动物细胞为模型的相关研究结论的普遍性以及植物细胞染色质构型的特征性,将在同一个水平上得到更为准确的验证,进一步促发人们对植物染色质构型的深入研究。
[1] Tiang CL, He Y, Pawlowski WP. Chromosome organization and dynamics during interphase, mitosis, and meiosis in plants., 2012, 158(1): 26–34.
[2] Lysak MA, Fransz PF, Ali HBM, Schubert I. Chromosome painting in., 2001, 28(6): 689–697.
[3] Bolzer A, Kreth G, Solovei I, Koehler D, Saracoglu K, Fauth C, Müller S, Eils R, Cremer C, Speicher MR, Cremer T. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes., 2005, 3(5): e157.
[4] Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, Politz JCR, Shendure J, Zhong S. The 4D nucleome project., 2017, 549(7671): 219–226.
[5] Doğan ES, Liu C. Three-dimensional chromatin packing and positioning of plant genomes., 2018, 4(8): 521.
[6] Bonev B, Cavalli G. Organization and function of the 3D genome., 2016, 17(11): 661–678.
[7] Lü HQ, Hao LL, Liu Y, Wu ZF, Han JQ. Current status and future perspectives in bioinformatical analysis of Hi-C data., 2020, 42(1): 87–99.吕红强, 郝乐乐, 刘源, 吴志芳, 韩九强. 基于生物信息学的Hi-C研究现状与发展趋势. 遗传, 2020, 42(1): 87–99.
[8] Ning CY, He MN, Tang QZ, Zhu Q, Li MZ, Li DY. Advances in mammalian three-dimensional genome by using Hi-C technology approach., 2019, 41(3): 215–233.宁椿游, 何梦楠, 唐茜子, 朱庆, 李明洲, 李地艳. 基于Hi-C技术哺乳动物三维基因组研究进展. 遗传, 2019, 41(3): 215–233.
[9] Sotelo-Silveira M, Montes RAC, Sotelo-Silveira JR, Marsch-Martínez N, de Folter S. Entering the next dimension: plant genomes in 3D., 2018, 23(7): 598–612.
[10] Liu C, Weigel D. Chromatin in 3D: progress and prospects for plants., 2015, 16(1): 170.
[11] Lieberman-Aiden E, Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of Long- Range interactions reveals folding principles of the human genome., 2009, 326(5950): 289–293.
[12] Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, Chew EGY, Huang PYH, Welboren WJ, Han YY, Ooi HS, Ariyaratne PN, Vega VB, Luo YQ, Tan PY, Choy PY, Wansa KDSA, Zhao B, Lim KS, Leow SC, Yow JS, Joseph R, Li HX, Desai KV, Thomsen JS, Lee YK, Karuturi RKM, Herve T, Bourque G, Stunnenberg HG, Ruan XA, Cacheux- Rataboul V, Sung WK, Liu ET, Wei CL, Cheung E, Ruan YJ. An oestrogen-receptor-α-bound human chromatin interactome., 2009, 462(7269): 58–64.
[13] Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation., 2002, 295(5558): 1306–1311.
[14] Zhang XY, He C, Ye BY, Xie DJ, Shi ML, Zhang Y, Shen WL, Li P, Zhao ZH. Optimization and quality control of genome-wide Hi-C library preparation., 2017, 39(9): 847–855.张香媛, 何超, 叶丙雨, 谢德健, 师明磊, 张彦, 沈文龙, 李平, 赵志虎. 全基因组染色质相互作用Hi-C文库制备的优化及其质量控制. 遗传, 2017, 39(9): 847–855.
[15] Zhao L, Wang SQ, Cao ZL, Ouyang WZ, Zhang Q, Xie L, Zheng RQ, Guo MR, Ma M, Hu Z, Sung WK, Zhang QF, Li GL. Chromatin loops associated with active genes and heterochromatin shape rice genome architecture for transcriptional regulation., 2019, 10(1): 1–13.
[16] Fraser J, Williamson I, Bickmore WA, Dostie J. An overview of genome organization and how we got there: from FISH to Hi-C., 2015, 79(3): 347–372.
[17] Zhu WS, Hu B, Becker C, Doğan ES, Berendzen KW, Weigel D, Liu C. Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecifichybrid., 2017, 18(1): 157.
[18] Grob S, Schmid MW, Grossniklaus U. Hi-C analysis inidentifies the, a structure with similarities to thelocus of., 2014, 55(5): 678–693.
[19] Feng SH, Cokus SJ, Schubert V, Zhai J, Pellegrini M, Jacobsen SE. Genome-wide Hi-C analyses in wild-type and mutants reveal high-resolution chromatin interactions in., 2014, 55(5): 694–707.
[20] Wang CM, Liu C, Roqueiro D, Grimm D, Schwab R, Becker C, Lanz C, Weigel D. Genome-wide analysis of local chromatin packing in., 2015, 25(2): 246–256.
[21] Zhang H, Zheng RQ, Wang YL, Zhang Y, Hong P, Fang YP, Li GL, Fang YD. The effects ofgenome duplication on the chromatin organization and transcriptional regulation., 2019, 47(15): 7857– 7869.
[22] Xie T, Zhang FF, Zhang HY, Wang XT, Hu JH, Wu XM. Biased gene retention during diploidization inlinked to three-dimensional genome organization., 2019, 5(8): 822–832.
[23] Wang MJ, Wang PC, Lin M, Ye ZX, Li GL, Tu LL, Shen C, Li JY, Yang QY, Zhang XL. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton., 2018, 4(2): 90–97.
[24] Wang MJ, Tu LL, Lin M, Lin ZX, Wang PC, Yang QY, Ye ZX, Shen C, Li JY, Zhang L, Zhou XL, Nie XH, Li ZH, Guo K, Ma YZ, Huang C, Jin SX, Zhu LF, Yang XY, Min L, Yuan DJ, Zhang QH, Lindsey K, Zhang XL. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication., 2017, 49(4): 579–587.
[25] Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, Radchuk V, Dockter C, Hedley PE, Russell J, Bayer M, Ramsay L, Liu H, Haberer G, Zhang X, Zhang Q, Barrero RA, Li L, Taudien S, Groth M, Felder M, Hastie A, Šimková H, Staňková H, Vrána J, Chan S, María MA, Ounit R, Wanamaker S, Bolser D, Colmsee C, Schmutzer T, Lala AS, Grasso S, Tanskanen J, Chailyan A, Sampath D, Heavens D, Clissold L, Cao S, Chapman B, Dai F, Han Y, Li H, Li X, Lin C, McCooke JK, Tan C, Wang P, Wang S, Yin S, Zhou G, Poland JA, Bellgard MI, Borisjuk L, Houben A, Doležel J, Ayling S, Lonardi S, Kersey P, Langridge P, Muehlbauer GJ, Clark MD, Caccamo M, Schulman AH, Mayer KFX, Platzer M, Close TJ, Scholz U, Hansson M, Zhang G, Braumann I, Spannagl M, Li C, Waugh R, Stein N. A chromosome conformation capture ordered sequence of the barley genome., 2017, 544(7651): 427–433.
[26] Dong PF, Tu XY, Li HX, Zhang JH, Grierson D, Li PH, Zhong SL. Tissue‐specific Hi‐C analyses of rice, foxtail millet and maize suggest non‐canonical function of plant chromatin domains., 2019. https://doi. org/10.1111/jipb.12809.
[27] Dong QL, Li N, Li XC, Yuan Z, Xie DJ, Wang XF, Li JN, Yu YN, Wang JB, Ding BX, Zhang ZB, Li CP, Bian Y, Zhang A, Wu Y, Liu B, Gong L. Genome-wide Hi-C analysis reveals extensive hierarchical chromatin interactions in rice., 2018, 94(6): 1141–1156.
[28] Dong PF, Tu XY, Chu PY, Lü PT, Zhu N, Grierson D, Du BJ, Li PH, Zhong SL. 3D chromatin architecture of large plant genomes determined by local A/B compartments., 2017, 10(12): 1497–1509.
[29] Zhou SL, Jiang W, Zhao Y, Zhou DX. Single-cell three- dimensional genome structures of rice gametes and unicellular zygotes., 2019, 5(8): 795–800.
[30] Peng Y, Xiong D, Zhao L, Ouyang WZ, Wang SQ, Sun J, Zhang Q, Guan PP, Xie L, Li WQ, Li GL, Yan JB, Li XW. Chromatin interaction maps reveal genetic regulation for quantitative traits in maize., 2019, 10(1): 2632.
[31] Boveri T. Die blastomerenkerne von ascaris megalocephala und die theorie der chromosomenindividualität., 1909, 3: 181–268.
[32] Manuelidis L, Borden J. Reproducible compartmentalization of individual chromosome domains in human CNS cells revealed by in situ hybridization and three-dimensional reconstruction., 1988, 96(6): 397–410.
[33] Armstrong SJ, Franklin FCH, Jones GH. Nucleolus- associated telomere clustering and pairing precede meiotic chromosome synapsis in., 2001, 114(23): 4207–4217.
[34] Fransz P, De Jong JH, Lysak M, Castiglione MR, Schubert I. Interphase chromosomes inare organized as well defined chromocenters from which euchromatin loops emanate., 2002, 99(22): 14584–14589.
[35] Liu C, Wang C, Wang GM, Becker C, Zaidem M, Weigel D. Genome-wide analysis of chromatin packing inat single-gene resolution., 2016, 26(8): 1057–1068.
[36] Liu C, Cheng YJ, Wang JW, Weigel D. Prominent topologically associated domains differentiate global chromatin packing in rice from., 2017, 3(9): 742–748.
[37] Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, Zhuang X. Spatial organization of chromatin domains and compartments in single chromosomes., 2016, 353(6299): 598–602.
[38] Fortin JP, Hansen KD. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data., 2015, 16(1): 180.
[39] Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, Yao X, Li F, Zhu W, Gao L, Chen H, Du X, Xie W, Xu X, Huang X, Liu J. 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis., 2017, 170(2): 367–381.
[40] Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, Haering CH, Mirny L, Spitz F. Two independent modes of chromatin organization revealed by cohesin removal., 2017, 551(7678): 51–56.
[41] Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions., 2012, 485(7398): 376–380.
[42] Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre., 2012, 485(7398): 381–385.
[43] Szabo Q, Jost D, Chang JM, Cattoni DI, Papadopoulos GL, Bonev B, Sexton T, Gurgo J, Jacquier C, Nollmann M, Bantignies F, Cavalli G. TADs are 3D structural units of higher-order chromosome organization in., 2018, 4(2): eaar8082.
[44] Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang XW. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells., 2018, 362(6413): eaau1783.
[45] Hong S, Kim D. Computational characterization of chromatin domain boundary-associated genomic elements., 2017, 45(18): 10403–10414.
[46] Yu M, Ren B. The three-dimensional organization of mammalian genomes., 2017, 33: 265–289.
[47] Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three- dimensional folding and functional organization principles of thegenome., 2012, 148(3): 458–472.
[48] Wutz G, Várnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, Walther N, Koch B, Kueblbeck M, Ellenberg J, Zuber J, Fraser P, Peters JM. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins., 2017, 36(24): 3573–3599.
[49] Lazar NH, Nevonen KA, O’Connell B, McCann C, O’Neill RJ, Green RE, Meyer TJ, Okhovat M, Carbone L. Epigenetic maintenance of topological domains in the highly rearranged gibbon genome., 2018, 28(7): 983–997.
[50] Dixon JR, Gorkin DU, Ren B. Chromatin domains: the unit of chromosome organization., 2016, 62(5): 668–680.
[51] Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T. High-resolution TADs reveal DNA sequences underlying genome organization in flies., 2018, 9(1): 189.
[52] Zhan YX, Mariani L, Barozzi I, Schulz EG, Blüthgen N, Stadler M, Tiana G, Giorgetti L. Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes., 2017, 27(3): 479–490.
[53] Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H,Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler S, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions., 2015, 161(5): 1012–1025.
[54] Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of chromosomal domains by loop extrusion., 2016, 15(9): 2038–2049.
[55] Levine M, Cattoglio C, Tjian R. Looping back to leap forward: transcription enters a new era., 2014, 157(1): 13–25.
[56] Gibcus JH, Dekker J. The hierarchy of the 3D genome., 2013, 49(5): 773–782.
[57] Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data., 2013, 14(6): 390–403.
[58] Chung IM, Ketharnathan S, Kim SH, Thiruvengadam M, Rani MK, Rajakumar G. Making sense of the tangle: insights into chromatin folding and gene regulation., 2016, 7(10): 71.
[59] Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping., 2014, 159(7): 1665–1680.
[60] Weber B, Zicola J, Oka R, Stam M. Plant enhancers: a call for discovery., 2016, 21(11): 974–987.
[61] Rodriguez-Granados NY, Ramirez-Prado JS, Veluchamy A, Latrasse D, Raynaud C, Crespi M, Ariel F, Benhamed M. Put your 3D glasses on: plant chromatin is on show., 2016, 67(11): 3205–3221.
[62] Cavalli G, Misteli T. Functional implications of genome topology., 2013, 20(3): 290.
[63] Denker A, De Laat W. The second decade of 3C technologies: detailed insights into nuclear organization., 2016, 30(12): 1357–1382.
[64] Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao HL, Stephens RM, Harris TJR, Munroe DJ, Wu XL. Long-range enhancers on 8q24 regulate c-Myc., 2010, 107(7): 3001–3005.
[65] Acemel RD, Maeso I, Gómez‐Skarmeta JL. Topologically associated domains: a successful scaffold for the evolution of gene regulation in animals., 2017, 6(3): e265.
[66] Crevillén P, Sonmez C, Wu Z, Dean C. A gene loop containing the floral repressor FLC is disrupted in the early phase of vernalization., 2013, 32(1): 140–148.
[67] Ariel F, Jegu T, Latrasse D, Romero-Barrios N, Christ A, Benhamed M, Crespi M. Noncoding transcription by alternative RNA polymerases dynamically regulates an auxin-driven chromatin loop., 2014, 55(3): 383–396.
[68] Hsieh THS, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. Mapping nucleosome resolution chromosome folding in yeast by micro-C., 2015, 162(1): 108–119.
[69] Bi X, Cheng YJ, Hu B, Ma X, Wu R, Wang JW, Liu C. Nonrandom domain organization of thegenome at the nuclear periphery., 2017, 27(7): 1162–1173.
[70] Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, Meyer BJ. Condensin- driven remodelling of X chromosome topology during dosage compensation., 2015, 523(7559): 240–244.
Progresses in the plant 3D chromatin architecture
Qianli Dong, Jinbin Wang, Xiaochong Li, Lei Gong
Chromatin architecture involves the patterns of chromatin coiling and packing as well as the mutual relative allocations of different chromatins. Besides the canonical microscopic observations, the chromatin architectural capture techniques, including the Hi-C and ChIA-PET, have been widely applied in characterization of chromatin architecture in various plant and animal model species, in which chromatin architectural features, such as the chromosome territory, compartment A/B, topological associated domains (TADs) and chromatin loops, were defined. As for the studies in plant species, replying on the two techniques above (with differences in experimental techniques and data structures), scientists have compared the variation of specific chromatin architecture features across species and/or in different cell types of the same plant species, besides detailed analyses in each individual model. Here, we mainly review the recent progresses in studies of plant chromatin architectures, in which their composition, establishing mechanism and effective factors were described and discussed. We also propose the main technical bottlenecks, describe the breaking-through progresses, and anticipate future research directions, which may offer more theoretical references for related researches in the field.
chromatin architecture; high-throughput chromosome conformation capture (Hi-C); chromatin interaction analysis by paired-end tag sequencing (ChIA-PET); compartment A/B; topological associated domains (TAD)
2019-10-29;
2019-12-19
国家重点研发计划项目(编号:2016YFD0101004),国家自然科学基金项目(编号:31670220)和2015“长白山学者计划”特聘教授专项资助[Supported by the National Key Research and Development Program of China (No. 2016YFD0101004), the National Natural Science Foundation of China (No. 31670220) and the Program of Changbai Mountain Scholar]
董芊里,博士研究生,专业方向:遗传学。E-mail: dongql043@nenu.edu.cn
王金宾,博士研究生,专业方向:遗传学。E-mail: wangjb702@nenu.edu.cn
李晓宠,博士研究生,专业方向:遗传学。E-mail: lixc800@nenu.edu.cn
董芊里、王金宾和李晓宠并列第一作者
宫磊,博士,教授,研究方向:进化生物学,基因组学和表观基因组学。E-mail: gongl100@nenu.edu.cn
10.16288/j.yczz.19-326
2020/1/2 18:15:51
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20191231.1147.004.html
(责任编委: 张飞雄)
