胃癌关键基因和通路的生物信息学和功能分析*
2020-03-04吴茜宋兴勃钟慧钰温阳应斌武
吴茜,宋兴勃,钟慧钰,温阳,应斌武
610041 成都,四川大学华西医院 实验医学科
胃癌的发病率较高,在癌症中排名前五,也是癌症相关死亡的三大主要原因之一,其中约有50%的死亡报告来自于东亚国家,如中国和日本[1]。虽然80%~90%的肿瘤发生是随机的,但仍然有研究证明遗传因素与胃癌的发生相关[2]。近年来,肿瘤生物学研究不断发展,胃癌的临床诊疗也随之进步,但由于胃癌发现时间一般较晚,造成临床诊疗延误,因而其预后效果仍不令人满意,胃癌患者的5年生存率较低,仅为25.1%[3-4]。相关学者针对胃癌的发病机制做了大量研究,许多基因被认为和胃癌的发生发展有关[5-6],但胃癌的发病机制仍旧没有达成统一的观点。确定胃癌早期诊断的相关指标和治疗胃癌的关键作用靶点十分重要,但相关方面的研究并不成熟。近年来,基因芯片和RNA测序等高通量技术的不断成熟使许多基因表达的相关数据得以呈现,计算机技术结合生物信息学的方法开始广泛应用于肿瘤研究,其在寻找肿瘤的诊断和靶向治疗的新型标志物方面的有效性和可靠性已经得到了证明。
本文在研究中拟选取基因表达综合数据库GEO(Gene Expression Omnibus)数据库中的GSE79973以及GSE19826数据集,将生物信息学方法应用到差异基因筛选和功能分析中,从而构建基因网络图,完成关键基因靶点的筛选,以期为探索胃癌的发病机制提供新的见解,从而为胃癌的早期诊疗提供帮助。
1 材料和方法
1.1 数据来源
在隶属于美国国立生物技术信息中心的GEO数据库中使用“gastric cancer”关键词,获得GSE79973和GSE19826基因表达谱,分别由He等[7]和Wang等[8]上传,均基于GPL570平台[HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array。共包含47例样本基因芯片数据,其中病例组(胃癌组织)22例,对照组(正常胃组织)25例。
1.2 数据分析流程
将基因芯片数据导入GCBI (Gene-Cloud of Biotechnology Information)分析平台进行数据分析。首先,根据数据的样本特征将其分为数据组和对照组,进行差异基因的筛选,随后对其进行基因本体(gene ontology,GO)分析、Pathway分析及在Pathway分析的基础上进行Pathway网络分析。最后,取GO分析和Pathway分析的交集结果进行基因信号通路网络分析及共表达网络分析。研究设计流程图如图1所示。
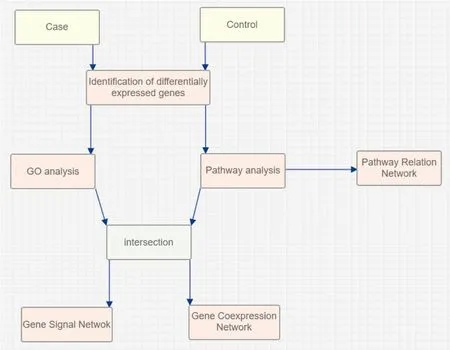
图1 研究设计流程图
Figure1.FlowDiagramofStudyDesign
1.3 差异基因筛选
基于GCBI平台并对基因芯片数据进行标准处理以利于分析并查看质控是否合格,在质控合格后设置参数(Q值<0.05,差异倍数>2)进行差异基因的筛选得到差异表达的基因(differentially expressed genes, DEGs)。
1.4 GO分析
GO广泛应用于生物信息学领域中,可分为“生物过程”、“分子功能”以及“细胞组分”三个部分,GO数据库可对基因产物进行简单注释,通常,GO注释之后的步骤是GO富集分析,富集的显著性用P值表示,针对富集的显著性设定一个阈值,用于区分该富集是否显著。利用GO富集分析可以寻找差异基因富集在在哪些途径、细胞定位、生物学功能中[9]。本研究设定错误发现率(false discovery rate,FDR)<0.05,利用GCBI平台对差异表达基因进行GO分析,并筛选出差异最显著的10个基因功能进行排序及分析。
1.5 Pathway 分析
Pathway分析是指代谢通路分析,通过对差异表达基因的Pathway显著性富集可以对其发挥最主要作用的代谢及信号传输途径进行了解。KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是进行基因产物在细胞中的功能及其代谢途径分析的数据库。本研究使用Fisher精确检验,设定P<0.05,基于GCBI平台对差异表达基因进行pathway分析,从而筛选出差异最显著的10个功能并对其按照显著性进行排序及分析。
1.6 Pathway关系网络分析
在KEGG数据库中,信号通路之间存在着上下游关系,根据此关系的不同,GCBI平台构建了利用交互网络图进行pathway研究的分析方法。基于pathway分析结果,显著性pathway之间的信号传导关系以直观网络图的方式进行呈现,通过网络图可以确定最上游具有调控作用的信号通路和网络中处于最下游的效应信号通路。通过对于pathway之间的关系分析,可以对信号传递过程有更深刻的认识。
1.7 共表达网络分析
大量研究表明,基因在表达过程中相互之间存在影响,这种相互作用关系使得基因表达和调控过程形成了一个巨大而复杂的网络。GCBI平台通过差异基因组成的表达谱数据进行基因共表达网络的构建。基因间的相互关系是具有层次性的,共表达网络可以将其清晰展现出来从而深入发现关键调控基因及其并呈现其相互作用的脉络。在共表达网络图中,根据基因之间调控关系的复杂程度,将每个基因用大小不同的点表示,点越大则说明调控关系更加复杂,即其所在位置在网格分析中的价值更高。
1.8 基因信号通路网络分析
基因信号网络对KEGG数据库进行了解构,突破了只能在单一pathway中获取基因表达过程中相互作用关系的局限。因而其可以在整个KEGG途径数据库中,寻找出某个蛋白质的上游或下游蛋白质。我们使用GCBI平台进行了基因信号通路网络分析,以确定关键通路和基因。
2 结 果
2.1 DEGs筛选结果
DEGs的识别是筛选样本间的高通量遗传数据具有显著差异的基因的统计方法。在数据预处理之后,与对照组织相比,在胃癌组织中筛选出共1 206个基因差异表达。在这些DEGs中,542个基因表达下调,664个基因表达上调。结果展示在火山图和聚类图中(图2和图3),可见DEGs能较好的区分出2个组。差异表达最显著的20个基因为GKN2、GKN1、ATP4B、GIF、LIPF、PGA3、ATP4A、PGC、AQP4、INHBA、ESRRG、DPCR1、KCNE2、KCNJ16、VSIG1、FUT9、CHIA、DPCR1、PGC和MFSD4(表1)。

图2 DEGs差异火山图
Figure2.VolcanoPlotofDEGs
Orange represents DEGs; the down-regulated DEGs are on the left side of the midline, and the up-regulated DEGs are on the right. DEGs: Differentially expressed genes.
2.2 GO分析结果
在本文中,使用GCBI平台获得的DEGs为GO富集分析形成了基础,计算GO的富集程度,展示了最可能与DEG相关的10个生物过程,包括:细胞粘附、细胞外基质组织、细胞外基质分解、胶原蛋白分解代谢过程、胶原纤维组织、小分子代谢过程、消化、血液凝固、血管生成、调节细胞增殖(表2)。
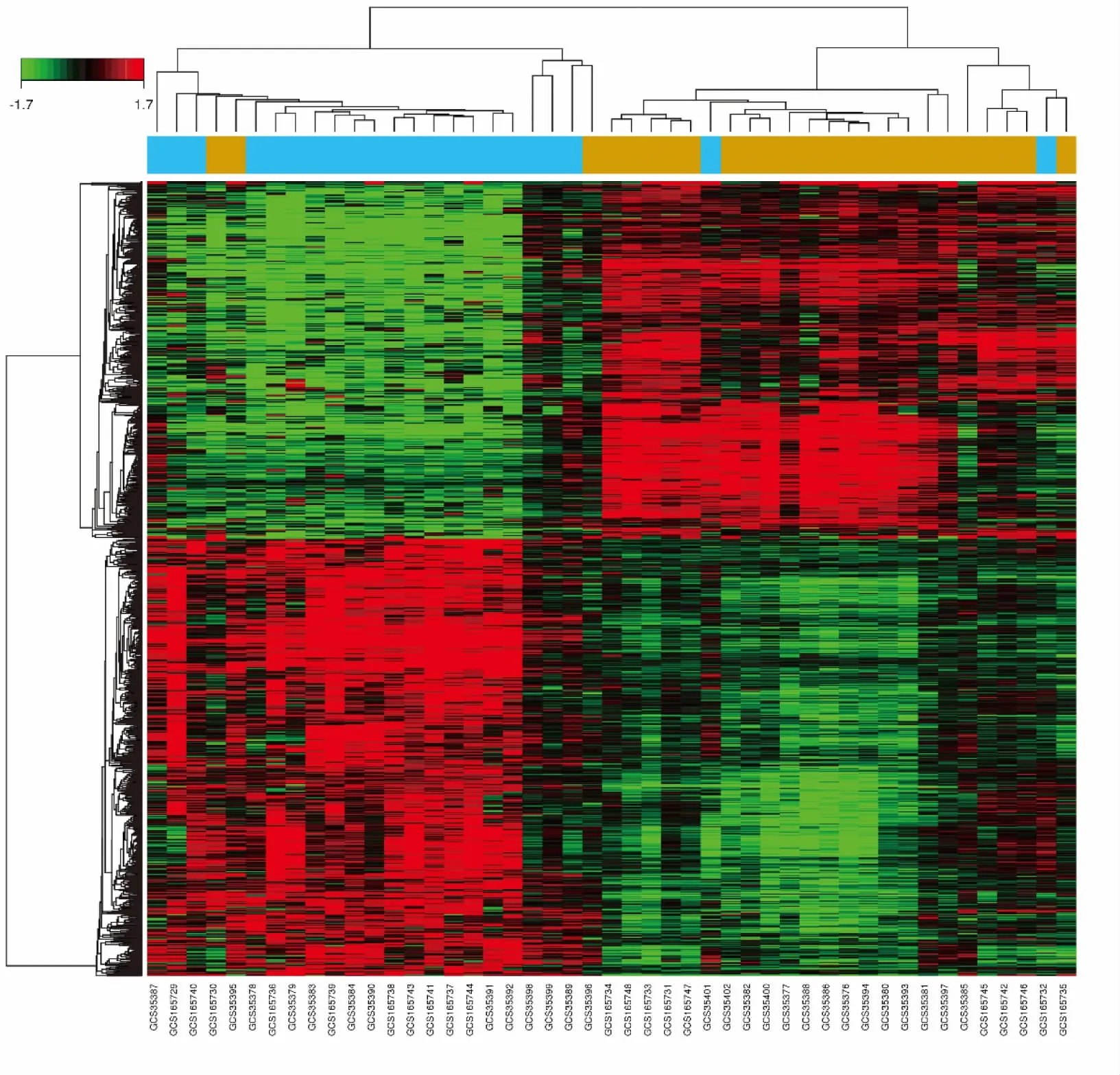
图3 DEGs聚类图
Figure3.DendrogramofDEGs
Blue indicates the gastric cancer group; Yellow indicates the control group; Red and green show differential gene expression in grouped samples; Red indicates that the expression value is high; Green indicates that the expression value is low.
表1 差异表达基因(前20个)
Table 1. Differentially Expressed Genes (Top 20)

No.GenesymbolAccession numberGene descriptionFold changePGene feature1GKN2NM_182536Gastrokine 2-97.35 <0.001down2GKN1NM_019617Gastrokine 1-94.05 <0.001down3ATP4BNM_000705ATPase, H+/K+ exchanging, beta polypeptide-67.70 <0.001down4GIFNM_005142Gastric intrinsic factor (vitamin B synthesis)-59.69 <0.001down5LIPFNM_001198828Lipase, gastric-59.33 <0.001down6PGA3NM_001079807Pepsinogen 3, group I (pepsinogen A)-57.18 <0.001down7ATP4ANM_000704ATPase, H+/K+ exchanging, alpha polypeptide-40.43 <0.001down8PGCNM_001166424Progastricsin (pepsinogen C)-26.69 <0.001down9AQP4NM_001650Aquaporin 4-26.07 <0.001down10INHBANM_002192Inhibin, beta A24.39 <0.001up11ESRRGNM_001134285Estrogen-related receptor gamma-22.13 <0.001down12DPCR1NM_080870Diffuse panbronchiolitis critical region 1-21.79 <0.001down13KCNE2NM_172201Potassium voltage-gated channel, Isk-related family, member 2-19.64 <0.001down
(Table 1 continues on next page)
(Continued from previous page)

No.GenesymbolAccession numberGene descriptionFold changePGene feature14KCNJ16NM_001270422Potassium inwardly-rectifying channel, subfamily J, member 16-19.08 <0.001down15VSIG1NM_001170553V-set and immunoglobulin domain containing 1-17.89 <0.001down16FUT9NM_006581Fucosyltransferase 9 (alpha (1,3) fucosyltransferase)-17.16 <0.001down17CHIANM_001040623Chitinase, acidic-16.64 <0.001down18DPCR1NM_080870Diffuse panbronchiolitis critical region 1-16.53 <0.001down19PGCNM_001166424Progastricsin (pepsinogen C)-16.22 <0.001down20MFSD4NM_181644Major facilitator superfamily domain containing 4-15.34 <0.001down
表2 差异表达基因GO分析
Table 2. GO Analysis of Differentially Expressed Genes

GO IDGO nameGene countsEnrichment scorePFDRGO:0007155Cell adhesion647.31<0.001<0.001GO:0030198Extracellular matrix organization4611.6<0.001<0.001GO:0022617Extracellular matrix disassembly2617.07<0.001<0.001GO:0030574Collagen catabolic process2417.28<0.001<0.001GO:0030199Collagen fibril organization1722.60<0.001<0.001GO:0044281Small molecule metabolic process772.93<0.001<0.001GO:0007586Digestion1716.03<0.001<0.001GO:0007596Blood coagulation414.57<0.001<0.001GO:0001525Angiogenesis256.45<0.001<0.001
GO: Gene ontology; FDR: False discovery rate.
2.3 Pathway分析及网络分析结果
通过对DEGs的pathway分析,我们可以确定富集DEGs的pathway项目,并确定可能与不同样本的DEGs相关的细胞通路变化,选择显著性最高的10条通路列表如表3。在网络分析中,信号通路具有更多的相互作用,因此其重要性更高。我们确定了共83条差异调节的pathway。这些途径包括ECM-受体相互作用、蛋白质消化吸收、胃酸分泌、PI3K-Akt信号通路等。在通路网络分析中,共筛选出来38条通路,其中包括3条上调通路,2条下调通路和33条上调/下调通路,寻找到38条通路中的71种关系(图4),其中粘附力、糖酵解/糖异生、Wnt信号通路、癌症的通路与上游和下游的相互作用较强。
表3 差异表达基因Pathway分析
Table 3. Pathway Analysis of Differentially Expressed Genes
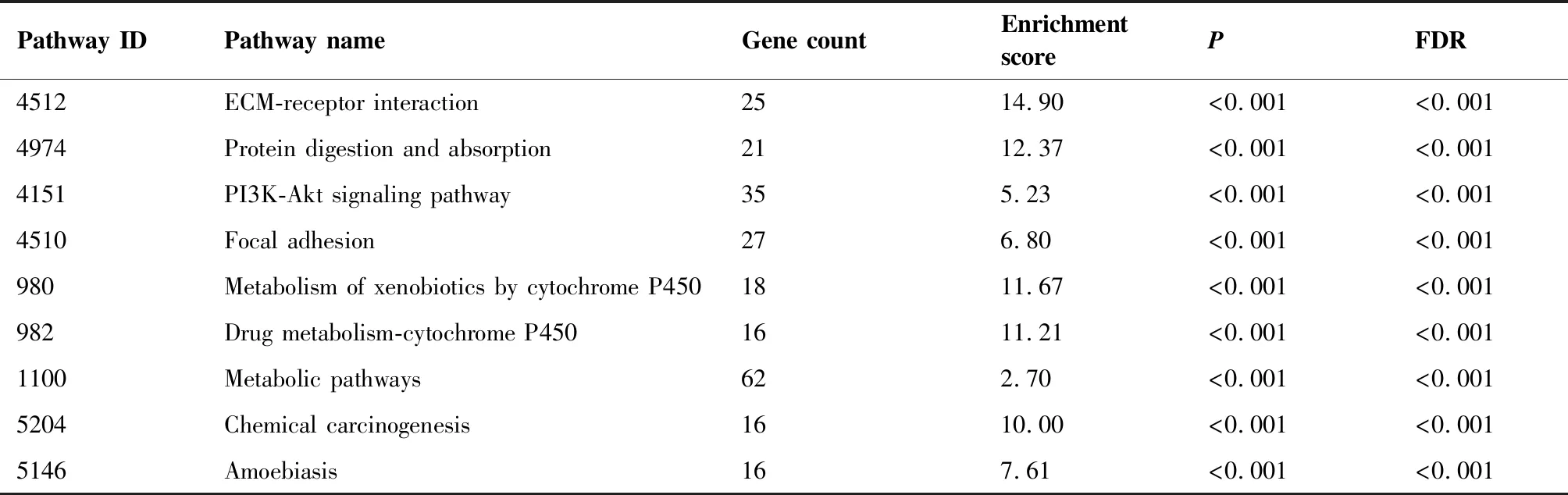
Pathway IDPathway nameGene countEnrichment scorePFDR4512ECM-receptor interaction2514.90 <0.001<0.0014974Protein digestion and absorption2112.37 <0.001<0.0014151PI3K-Akt signaling pathway355.23 <0.001<0.0014510Focal adhesion276.80 <0.001<0.001980Metabolism of xenobiotics by cytochrome P4501811.67 <0.001<0.001982Drug metabolism-cytochrome P4501611.21 <0.001<0.0011100Metabolic pathways622.70 <0.001<0.0015204Chemical carcinogenesis1610.00 <0.001<0.0015146Amoebiasis167.61 <0.001<0.001
FDR: False discovery rate; ECM: Extracellular matrix.
2.4 共表达网络分析及信号通路网络分析结果
我们挑选出来自GO和Pathway分析的263个重叠基因,将它们应用于基因共表达网络构建和信号通路网络分析。共表达网络分析图如图5所示,共46个节点,代表136个基因,分布以聚集性分布呈现,符合共表达理论。其中前10 个基因的信息如表4所示。信号通路网络分析共有123个中枢基因,212个关系,前10个中枢基因的信息如表5所示。

图4 Pathway网络分析图
Figure4.PathwayandNetworkAnalysis
Red represents up-regulation; Blue represents down-regulation; Yellow represents up-and down-regulation. The larger the points are, the more signal paths with upstream and downstream interactions are.
表4 差异表达基因共表达分析
Table 4. Co-Expression Analysis of Differentially Expressed Genes

No.Gene symbolDegreeFunctionDescription1SH3GL223CodingSH3-domain GRB2-like 22CKMT218CodingCreatine kinase, mitochondrial 2 (sarcomeric)3CHIA14CodingChitinase, acidic4ATP4A14CodingATPase, H+/K+ exchanging, alpha polypeptide5CCKBR14CodingCholecystokinin B receptor6TPCN214CodingTwo pore segment channel 27ATP4B13CodingATPase, H+/K+ exchanging, beta polypeptide8KCNE213CodingPotassium voltage-gated channel, Isk-related family, member 29CLCNKB11CodingChloride channel, voltage-sensitive Kb10KCNJ1610CodingPotassium inwardly-rectifying channel, subfamily J, member 16
表5 差异表达基因信号通路分析
Table 5. Analysis of Differentially Expressed Genes and Signaling Pathways

No.Gene symbolGene descriptionBetweennessIndegreeOutdegree1UGT2B15UDP glucuronosyltransferase 2 family, polypeptide B15793.912122ITGA2integrin, alpha 2 (CD49B, alpha 2 subunit of VLA-2 receptor)351.52223ITGB1integrin, beta 1 (fibronectin receptor, beta polypeptide, antigen CD29 includes MDF2, MSK12)351.52224CYP3A4cytochrome P450, family 3, subfamily A, polypeptide 496.711115CYP3A5cytochrome P450, family 3, subfamily A, polypeptide 596.711116GSTA1glutathione S-transferase alpha 113.211117GSTA3glutathione S-transferase alpha 313.211118GSTA4glutathione S-transferase alpha 413.211119ALDH1A1aldehyde dehydrogenase 1 family, member A177.89910PRKXprotein kinase, X-linked894.044
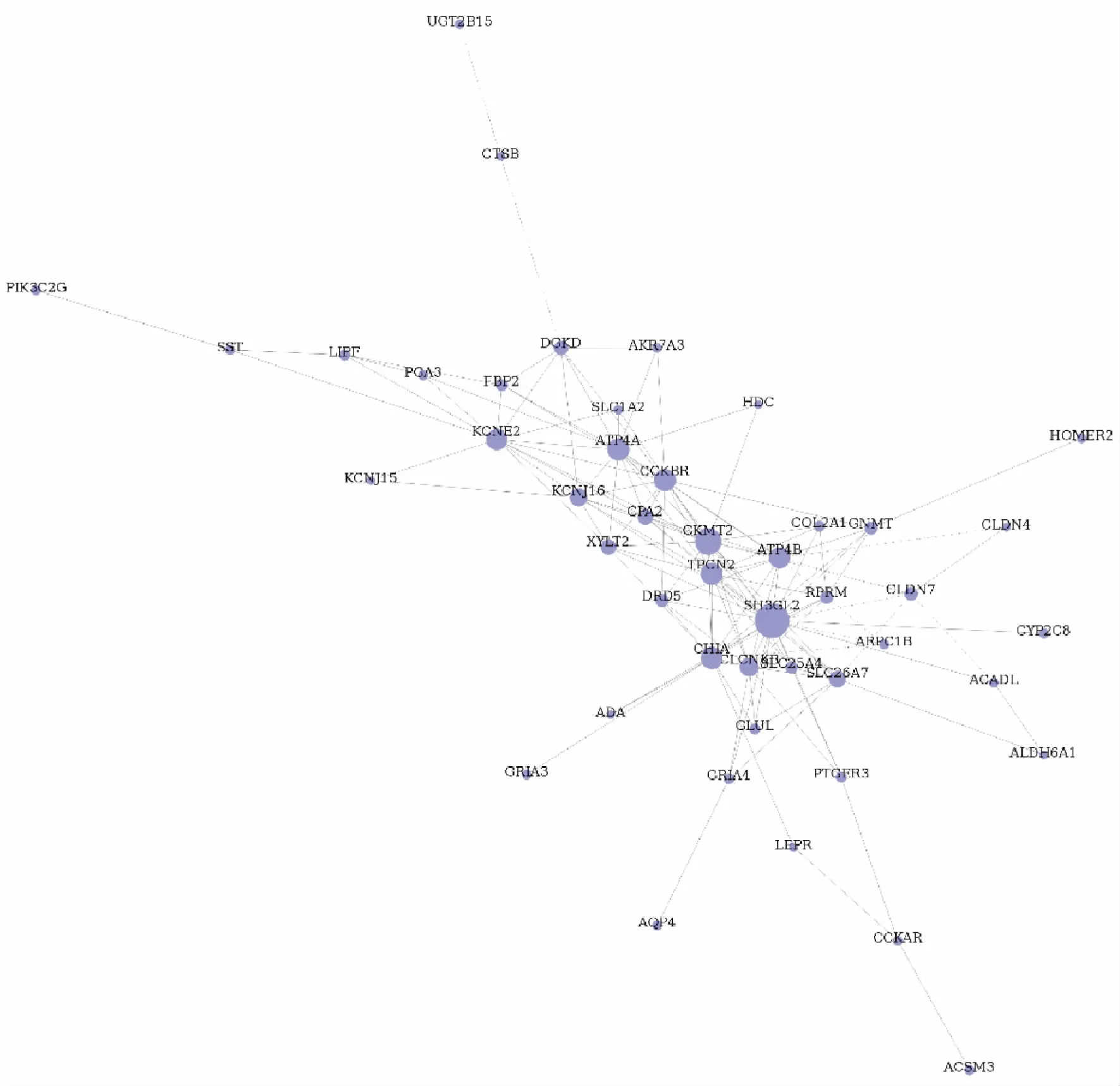
图5 差异表达基因共表达网络图
Figure5.Co-ExpressingNetworkofDifferentiallyExpressedGenes
Solid lines indicate positive correlations, and dashed lines indicate negative correlations. The larger the point is, the more genes it interacts with.
3 讨 论
近年来,大量研究表明分子生物标志物在疾病诊断、预后预测和靶向治疗中发挥重要作用。随着诊疗水平的不断进步,虽然过去的一个世纪里胃癌的发病率有所下降,但在世界范围内胃癌仍然是危害极大的一种重大疾病。幽门螺旋杆菌感染、饮食不当、卫生条件落后和吸烟是常见的危险因素[10]。另外,胃癌的诊断延误和转移扩散是胃癌死亡的主要原因。因此,寻找新的分子标志物对胃癌的早期诊断,针对性治疗和预后评估至关重要。
我们使用生物信息学分析筛选了涉及胃癌发生发展的DEGs。结果表明,与正常组织相比,胃癌组织中数千个基因发生了变化,本研究共确定了1 206个DEGs,包括664个上调基因和542个下调基因。我们对其进行了进一步的筛选,找出了具有显著表达变化的前20个基因,其中GKN2和GKN1的表达明显下调。Dokhaee等[11]纳入27例胃癌组织样本,通过Real-time PCR对GKN2和GKN1进行定量分析,结果表明与正常组织相比,癌症标本中GKN1和GKN2基因表达显著降低(P值分别为0.008,0.004)。另外,Song等[12]通过高通量计算分析发现GKN2在胃癌组织中明显下调,后在细胞实验中验证了GKN2在胃癌体外增殖和侵袭中具有抗癌作用的具体机制,即GKN2的体外过度表达使PTEN/PI3K/AKT/mTOR和JAK/STAT通路沉默,进而抑制胃癌增殖和侵袭,这与本文章的预测结果一致。具有显著表达变化的前20个基因中,INHBA表达明显上调,据报道INHBA表达水平在癌组织中显著高于在邻近正常粘膜中的表达水平,并且与低表达病例相比,INHBA高表达与较差的5年存活率相关,INHBA被证实是一个独立的预后因素[13]。
GO富集分析显示,DEGs主要参与细胞粘附、细胞外基质、细胞外基质分解、胶原分解代谢、胶原纤维、血管生成等方面。Pathway分析发现,差异最显著的富集通路主要涉及细胞外基质-受体相互作用、蛋白质消化吸收、PI3K-Akt信号通路、细胞粘附、代谢途径、癌症通路等。将基因间相互作用形成网络后可见,胃癌组织和正常组织之间的差异也主要集中在细胞粘附、Wnt信号通路、癌症通路等方面,以此可推测胃癌的发生发展与细胞外基质、细胞粘附、癌症通路的相关性较大。
基于GCBI平台的信号通路网络分析解构了KEGG数据库,突破了KEGG-Pathway数据库中基因间相互作用关系局限于某Pathway的限制,是在全KEGG-Pathway数据库的范围内筛选某个蛋白的上游或下游蛋白。差异表达基因的信号通路相互作用分析显示,UGT2B15作为中枢基因,上下游各与12个基因有联系,该基因涉及的功能主要有外源性化合物代谢过程、细胞葡萄糖醛酸化、类固醇代谢过程、黄酮类生物合成过程。葡萄糖醛酸化是消除外源性和内源性化合物(如环境致癌物质和体内雄激素)的主要途径,有研究表明,UGT2B15的基因多态性与前列腺癌[14]和胃癌[15]的发生发展有关,可能是致癌基因,并且是癌症的治疗靶标。ITGA2和ITGB1均有22个上游基因和2个下游基因,属于整合素家族,整合素属于细胞粘附受体家族成员,是一类广泛分布于细胞表面的跨膜糖蛋白,多项研究证实整合素涉及多个肿瘤的侵袭转移,如Lahlou等[16]通过转基因小鼠肿瘤模型提供了较为明确的ITGB1在乳腺癌细胞侵袭与转移中起重要作用的体内实验证据。Chuang等[17]发现ITGA2不仅抑制胃癌细胞迁移,还诱导胃癌细胞的凋亡,并且ITGA2的较高表达水平在抗ITGA2抗体治疗期间对凋亡进展的有着显著影响。GSTA1、GSTA3和GAST4均有11个上游基因和11个下游基因,属于GST基因家族,GST基因家族通过还原型谷胱甘肽与许多底物(如药物和环境污染物)的结合,编码对某些生命过程以及解毒和毒性机制至关重要的基因。GST基因在氧化应激反应中被上调,并且在许多肿瘤中会过度表达,增强或减弱机体对化疗药物的反应[18]。
共表达网络分析是通过差异基因组成的表达谱数据来构建基因共表达网络。共表达网络能够层次清晰地展现基因间的相互关系,更深入地发现关键调控基因以及相互作用脉络,可以揭示在健康状态和疾病状态之间存在差异的基因,并且有助于揭示潜在的疾病和其他表型调节因子,或揭示一些新的基因相互作用关系[19]。如Kong等[20]通过共表达网络分析确定了与肝细胞癌的进展和预后相关的五个核心基因。差异表达基因的共表达网络分析显示SH3GL2、CKMT2、CHIA、ATP4A、CCKBR、TPCN2分别有23、18、14、14、14、14个基因呈现共表达趋势,这些基因的作用主要体现在信号转导、物质代谢过程、炎症反应的介质分子、免疫应答、跨膜运输等方面。有文献报道,SH3GL2的敲除促进了胶质瘤细胞的迁移和侵袭,而SH3GL2的过表达则抑制了它们的迁移和侵袭。进一步的分子机制研究发现,SH3GL2的沉默明显激活了STAT3信号,从而促进了MMP2的表达和分泌。SH3GL2通过负向调控STAT3/MMP2信号通路抑制胶质瘤细胞的迁移和侵袭行为,SH3GL2的缺失可能会增强STAT3/MMP2信号通路,从而促进胶质瘤细胞的迁移和侵袭[21]。CKMT2是维持正常能量代谢时必不可少的,有研究表明CKMT2可能是参与骨肉瘤形成的关键调节因子[22]。因此,共表达网络分析得到的SH3GL2、CKMT2、CHIA、ATP4A、CCKBR、TPCN2等节点基因,或许是胃癌进展过程中的关键基因,但这主要是基于数据推测,需要进一步实验验证。
总之,我们目前的研究提供了一个全面的生物信息学分析,包括DEGs的筛选,以及对关注基因进行了GO分析、Pathway分析、基因信号通路网络分析、共表达分析,得到可能与胃癌发生、演进相关的基因。目前的结果可以促进我们对胃癌相关潜在分子机制的理解。例如INHBA、UGT2B15、ITGA2、ITGB1、SH3GL2等基因,及其相关的生物过程如细胞粘附、胶原纤维、细胞外基质-受体相互作用等,或可提供胃癌诊断和治疗的潜在靶标。
作者声明:本文全部作者对于研究和撰写的论文出现的不端行为承担相应责任;并承诺论文中涉及的原始图片、数据资料等已按照有关规定保存,可接受核查。
学术不端:本文在初审、返修及出版前均通过中国知网(CNKI)科技期刊学术不端文献检测系统的学术不端检测。
同行评议:经同行专家双盲外审,达到刊发要求。
利益冲突:所有作者均声明不存在利益冲突。
文章版权:本文出版前已与全体作者签署了论文授权书等协议。
