中国北方黑唇苜蓿盲蝽种群遗传多样性和遗传分化分析
2020-03-04王梦琦张利娟张宏瑞孙跃先
王梦琦, 张利娟, 张宏瑞, 孙跃先,*
(1.云南农业大学植物保护学院, 昆明 650201; 2.河南农业大学植物保护学院, 郑州 450002)
近年来,随着转基因棉花的大面积种植,用于防治棉铃虫Helicoverpaarmigera等鳞翅目害虫的广谱性杀虫剂使用频率降低,导致棉盲蝽、棉蓟马、棉蚜Aphisgossypii、烟粉虱Bemisiatabaci等次要害虫逐渐上升至主要害虫。黑唇苜蓿盲蝽Adelphocorisnigritylus属于半翅目(Hemiptera)盲蝽科(Miridae)苜蓿盲蝽属Adelphocoris(郑乐怡, 1998),是一种重要的棉花害虫。黑唇苜蓿盲蝽的若虫和成虫刺吸棉花的花序、花蕾、雌蕊、雄蕊,造成落花、落蕾、种子发育不全使棉花大量减产。同时,黑唇苜蓿盲蝽寄主范围广泛,除危害棉花外还危害苜蓿、马铃薯等作物,对农业生产造成严重危害。
群体遗传学是遗传学中的一个重要分支,常用于追溯害虫起源,分析种群历史动态等研究。了解物种的进化历史可以帮助我们预测物种应对环境变化的能力,并有利于物种的管理和保护。寄主和人为干扰是影响种群遗传结构和种群动态的两个重要因素,如Wei等(2015)在梨小食心虫Grapholitamolesta的研究中发现我国北方地区种群规模在末次盛冰期后有所扩大,这一结果可能与人类对其寄主植物的栽培活动有关。Cao等(2017)对西花蓟马Frankliniellaoccidentalis的研究表明,在中国西花蓟马种群的遗传分化是由多次引入和人为媒介的零星扩散导致的,与其地理分布无关。另外,环境气候也是影响种群动态的主要因素,Qin等(2018)对世界范围内橘小实蝇Bactroceradorsalis群体遗传的研究发现,在中国境内的橘小实蝇正向中部地区扩张,中国中部地区的气候与欧洲和北美温带地区类似并表明橘小实蝇能够在温带冬季生存。
线粒体DNA作为一种广泛使用的分子标记,常被用于开展群体遗传学研究(Aviseetal., 1987; Ballard and Pichaud, 2014),在中性进化假设下推断物种的进化关系,分析其群体历史动态。其中COI,COII,Cytb,ND4和ND5等蛋白质编码基因在群体遗传学研究中应用频率较高,如高立志(2014)采用ND4和微卫星分子标记对我国柑橘大实蝇Bactroceraminax18个地理种群的遗传结构进行研究,发现华南种群是其在我国的扩散中心,十堰种群被认定为是华南地区向华中地区扩散的桥头堡种群。Meng等(2018)使用COI,Cytb和ND5基因的串联序列分析柑橘木虱Diaphorinacitri群体遗传,发现我国种群的种内变异程度低,没有形成明显的群体聚类,并显示其近期发生了种群扩张,作者推测我国柑橘品种及种植面积大幅度增加是扩张发生的主要原因。综上,已有研究证实COI,COII,Cytb,ND4和ND5等基因片段可被有效地用于群体遗传学研究。
本研究通过对多个线粒体基因分子标记的筛选,最终选取线粒体ND5,ND4和Cytb基因片段用于中国北方地区黑唇苜蓿盲蝽群体遗传学研究,期望明确其遗传多样性、遗传结构等现状,为实现该害虫的综合防治提供一定的理论基础。
1 材料与方法
1.1 试虫
本研究所用的黑唇苜蓿盲蝽标本于2012-2016年期间采集自中国北方地区12个地理种群233头成虫,采用扫网法进行标本的收集,每个种群设置3个以上的取样点,各个取样点之间间隔1 000 m左右,同时考虑每个取样点小生境的差异性,例如寄主植物的种类组成等。样品收集信息见表1。采集标本均用95%酒精保存,并存放于-20℃冰箱中。

表1 黑唇苜蓿盲蝽样品收集信息
1.2 PCR扩增及测序
将黑唇苜蓿盲蝽成虫标本的腹部和翅去除,剩余组织作为提取DNA的实验材料,使用TIANamp Genomic DNA Kit提取DNA,而后存放于-20℃冰箱中备用。分别以黑唇苜蓿盲蝽线粒体基因组(GenBank登录号: NC_027144.1)ND5,ND4和Cytb基因序列为模板设计特异性引物表2,引物由上海生工生物公司合成。

表2 PCR引物
PCR反应体系(25 μL): DNA模板(50 ng/μL)2 μL, 正反向引物(10 mol/μL)各1 μL, 2×Taq PCR Master Mix 12.5 μL, ddH2O补足25 μL。PCR反应程序: 94℃预变性3 min; 94℃变性 1 min, 退火1 min(退火温度:ND5, 50~55℃;ND4, 58~60℃;Cytb, 45~50℃), 72℃延伸1 min, 共30个循环;72℃延伸5 min, 4℃保存。扩增产物使用1%琼脂糖凝胶电泳进行检测,产物纯化后送往上海生工生物公司完成双向测序。
1.3 数据分析
1.3.1种群遗传多样性分析:使用SeqMan(Thompsonetal., 1997; Larkinetal., 2007)软件进行正反向序列拼接。然后在GenBank进行在线BLAST比对以检验所得序列的准确性,通过MEGA 10.0(Kumaretal., 2018)使用默认参数进行序列比对,并将核苷酸序列翻译成氨基酸进行校正,检查序列中间是否有终止密码子及碱基的插入缺失出现;同时计算变异位点等信息以及核苷酸突变率。采用DnaSP 6.0(Rozasetal., 2017)软件统计单倍型数目(h)、单倍型多样性(Hd)、核苷酸多样性(π)、变异位点数(S)、平均核苷酸差异数(K)等参数。
1.3.2种群遗传结构分析:利用SAMOVA 1.0(Dupanloupetal., 2002)对不同地理种群间的空间变异进行分析,分组数从2依次增加到15,每次分组计算均进行100次的置换捡验。利用Arlequin 3.5(Excoffier and Lischer, 2010)分别计算种群间、种群内、组间和组内个体间的遗传变异情况,进行10 000次置换以检验显著性水平。利用Arlequin软件中的Tamura and Nei模型(Tamura and Nei, 1993)计算种群间成对的遗传分化指数(Fst)矩阵,并将其转换为Fst/1-Fst,利用各地理种群的经纬度信息计算种群间的直线地理距离(km)矩阵,将其转换为对数形式,通过NTSYSpc 2.10e(Rohlf, 1998)软件进行Mantel检验,即种群间遗传距离与地理距离的相关性。
1.3.3系统发育关系:通过MEGA 10.0以三点苜蓿盲蝽Adelphocorisfasciaticollis线粒体基因组序列(GenBank登录号:NC_023796.1)作为外群构建单倍型发育树,使用Kimura 2-Parameter模型,系统发育分支的置信度通过自展检验获得并进行1 000次重复检测。使用PopART 1.7软件(Leigh and Bryant, 2015),以中介网络(median-joining network)法构建单倍型网络关系图,推测各个单倍型间的进化关系。
1.3.4历史动态分析:采用DnaSP 6.0软件分别对己划分组和所有样本进行分析,计算Fu and Li’sF*和Fu and Li’sD*值(Fu and Li, 1993);利用Arlequin软件计算Fu’sFs和Tajima’sD值,通过这些参数检验种群是否偏离中性进化模型。使用Arlequin软件进行错配分布分析,推断种群是否发生扩张,同时对观测到的错配分布进行平滑指数和错配分布预测值和观测值间方差总和的计算。
2 结果
2.1 种群线粒体基因序列
本研究最终获得线粒体ND5,ND4和Cytb基因片段可用序列各233条(GenBank登录号: MT862031-MT862129)。单独分析各线粒体基因时,ND5基因序列长度为858 bp,检测出33个单倍型,单倍型多样性(Hd)为0.770;ND4基因序列长度为804 bp,检测出34个单倍型,单倍型多样性(Hd)为0.806;Cytb基因序列长度为816 bp,检测出32个单倍型,单倍型多样性(Hd)为0.780。无插入和缺失位点,排除假基因的可能后,将3个线粒体基因片段(ND5: 858 bp;ND4: 804 bp和Cytb: 816 bp)串联后进行综合分析, 串联序列长度为2 226 bp。在串联序列中共检测到141个多态性位点,占序列长度的5.69%,其中简约信息位点为88个,单一变异位点为53个。碱基组成分析发现,A=29.7%, T=45.9%, G=13.5%, C=10.9%,A+T含量为56.8%,明显大于G+C含量(43.2%),碱基组成表现出了明显的偏倚,符合昆虫线粒体基因序列的碱基组成特点。在串联序列中共检测到87个单倍型,其中Hap3被47个个体所共享,Hap9被35个个体所共享,Hap8被22个个体所共享。
2.2 种群遗传多样性
从串联序列分析可以看出(表3),12个黑唇苜蓿盲蝽种群均显示出较高的单倍型多样性和较低的核苷酸多样性,各个种群间存在一定的多态性差异:各种群序列变异位点数(S)范围为5~63,单倍型数目(h)范围为2~15,单倍型多样性(Hd)范围为0.596~0.939,核苷酸多样性(π)为0.00083~0.00687,平均核苷酸差异数(K)为2.067~17.013。河北衡水种群(HS)显示出相对较高的遗传多样性,其单倍型数目为15、核苷酸多样性为0.00687、平均核苷酸差异数为17.013。河北衡水种群(HS)、山东德州种群(DZ)、山东菏泽种群(HZ)单倍型多样性较高,分别为0.939, 0.937和0.933。山东德州种群(DZ)、河北衡水种群(HS)、辽宁铁岭种群(TL)、黑龙江绥化种群(SH)序列变异位点数最高,分别为63, 61, 58和54。山东潍坊种群(WF)单倍型多样性最低,为0.596,同时单倍型数目较少。

表3 基于线粒体基因ND5, ND4和Cytb串联序列的中国北方黑唇苜蓿盲蝽种群遗传多样性和种群历史动态参数
2.3 种群遗传结构
基于串联序列的分析,12个种群两两之间的Fst值为-0.06880~0.82926(表4)。河北廊坊种群(LF)与其余11个种群存在显著的遗传差异,山东德州种群(DZ)与另外6个种群的遗传分化较高。但陕西商洛种群(SL)、河北涿州种群(ZHZ)和河南郑州种群(ZZ)两两之间的Fst值较低,分别为-0.00249(SL-ZHZ),-0.00297(SL-ZZ)和0.00230(ZHZ-ZZ)。

表4 基于线粒体基因ND5, ND4和Cytb串联序列的中国北方黑唇苜蓿盲蝽种群间遗传分化指数(Fst)
SAMOVA分析结果如下(图1):当K=2到3时,组间遗传差异FCT值呈上升趋势,当K=3~6时呈缓慢下降的趋势;K=7~9,FCT值呈缓慢上升的趋势,组内种群间和种群内遗传差异值FSC与FST在K=2~9时呈明显下降趋势。结合种群间遗传差异分析结果和SAMOVA分析,作者选取K=5作为最佳分组数量,第1组包括河北廊坊种群(LF),第2组包括黑龙江绥化种群(SH),第3组包括北京朝阳种群(BJ)、吉林松原种群(SY)、辽宁铁岭种群(TL),第4组包括河北涿州种群(ZHZ)、河北衡水种群(HS)、河南郑州种群(ZZ)、山东淮坊种群(WF)、陕西商洛种群(SL),第5组包括山东德州种群(DZ)、山东菏泽种群(HZ)。组间遗传差异分析均达到显著水平(表5),其中,第1组与第5组间遗传差异最大,为0.60978(P<0.001);第1和第4组间的遗传差异较小,为0.25891(P<0.01)。SAMOVA分析结果显示组间的变异为17.09%,组内的变异为82.91%,表明黑唇苜蓿盲蝽的遗传分化主要表现为组内分化。Mantel检验结果显示种群间遗传距离与地理距离不存在显著的相关性(r=-0.02735,P=0.4310)。
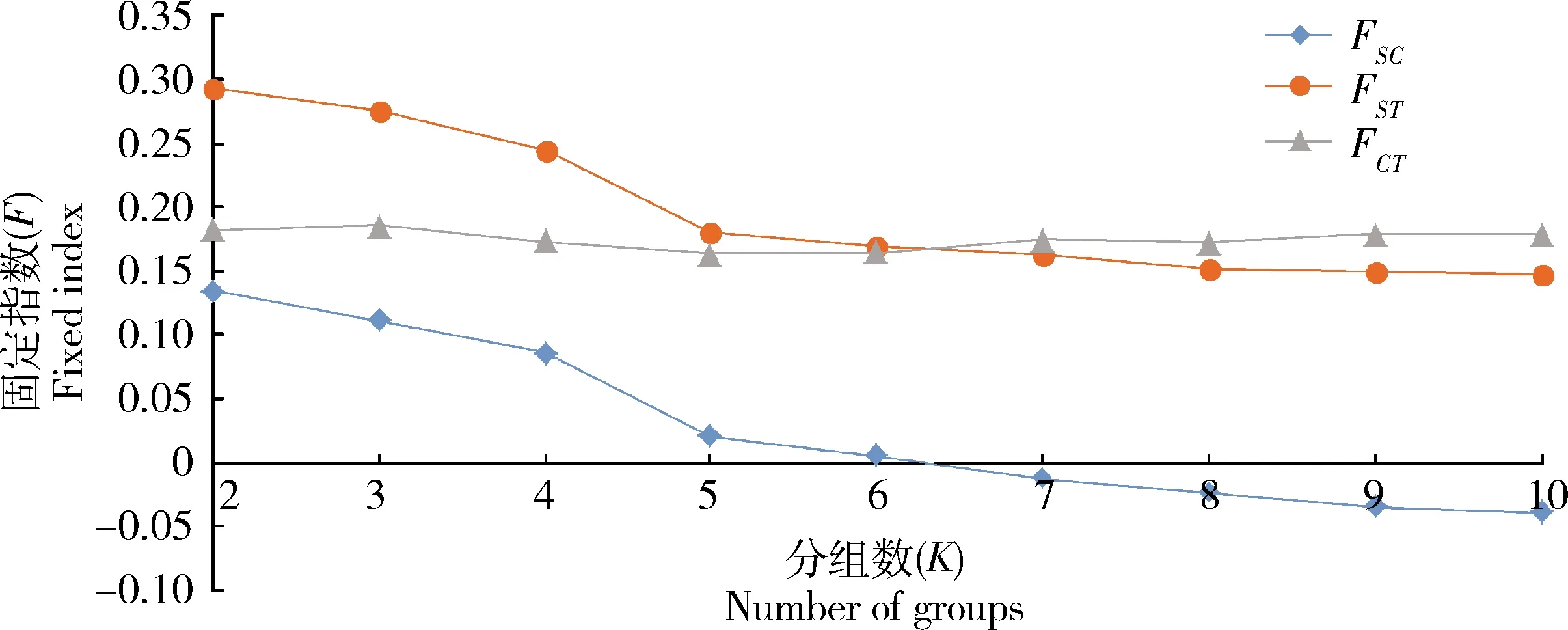
图1 基于线粒体基因ND5, ND4和Cytb串联序列的中国北方黑唇苜蓿盲蝽种群固定指数(F)随着分组组数(K)的变化

表5 基于线粒体基因ND5, ND4和Cytb串联序列的中国北方黑唇苜蓿盲蝽种群组间遗传分化指数(Fst)
2.4 单倍型系统发育关系
系统发育树显示,同一种群包含的单倍型并没有聚到同一支系中,而是相互混杂聚在一起,单倍型间许多属于多分歧分支,进化关系不明显(图2)。单倍型网络进化关系显示(图3),单倍型分化节点和共享频率迥异,其中Hap3, Hap5, Hap8, Hap9和Hap48位于单倍型网络图的中间位置,其余单倍型则是由原始单倍型通过一次或几次突变形成并分布在其他种群中,部分单倍型之间通过缺失的中间单倍型相互连接。在高频率共享单倍型中,Hap3被4个组群(除第2组)共享,Hap8和Hap9分别被第2, 3和4组及第3, 4和5组各3个组群所共享。该单倍型关系图与系统发育树结果一致,各单倍型相互散布在不同的地理种群中,未形成明显的系统地理格局。

图3 基于线粒体基因ND5, ND4和Cytb串联序列的中国北方黑唇苜蓿盲蝽种群单倍型间的进化网络图
2.5 种群历史动态
中性检验结果(表3)表明,将12个种群作为一个整体进行分析时,Fu and Li’sF*和Fu and Li’sD*为显著负值,而 Fu’sFs和Tajima’sD为负值但未达显著水平。当以组为单位进行分析时,第1组和第5组Tajima’sD, Fu and Li’sF*和Fu and Li’sD*均为显著负值,而 Fu’sFs为负值且未达显著水平;第3组的结果同整体分析一致;第4组Fu’sFs和Fu and Li’sF*和Fu and Li’sD*均为显著负值,而Tajima’sD为负值但未达显著水平。单独对种群进行分析发现,河北廊坊种群(LF)只有Fu’sFs为负值且不显著,其余3个参数均为显著负值;辽宁铁岭种群(TL)和山东淮坊种群(WF)有两个参数显著负值;山东德州种群(DZ)和山东菏泽种群(HZ)均有一个参数为显著负值。整体种群的错配分析(图4)呈单峰,SSD和HR指数均无统计学意义。

图4 基于线粒体基因ND5, ND4和Cytb串联序列的中国北方黑唇苜蓿盲蝽种群的错配分析图
3 讨论
遗传多样性是由核苷酸序列发生改变引起的,其中单倍型多样性(Hd)和核苷酸多样性(π)常被作为评价物种遗传多样性的重要指标,通常单倍型数、单倍型多样性指数和核苷酸多样性参数值越大,表明物种种群遗传多样性越高。本研究黑唇苜蓿盲蝽种群拥有高的单倍型多样性(Hd=0.924)和低的核苷酸多样性(π=0.00424),与已发表中黑盲蝽Adelphocorissuturalis、烟盲蝽Nesidiocoristenuis和黑唇苜蓿盲蝽遗传多样性结果(Zhangetal., 2015; Xunetal., 2016; 张利娟等, 2018)相似。按照Grant和Bowen(1998)提出的基于单倍型多样性(Hd)和核苷酸多样性(π)推测种群进化历史的标准,通常Hd≥0.5且π<0.5%时,是由于种群经历瓶颈效应后迅速膨胀所致。作者推测出现上述现象的主要原因是由于黑唇苜蓿盲蝽种群近期呈现快速增长的模式,核苷酸突变累积时间不足造成的(Frankham, 1996; Cooketal., 1997;Grant and Bowen, 1998)。种群间遗传分化指数(Fst)显示黑唇苜蓿盲蝽组间遗传分化程度较低,Mantel检验结果显示遗传距离和地理距离两者不具有相关性。这一结果说明我国不同地理与气候条件不是影响黑唇苜蓿盲蝽遗传分化的主要因素。整体上黑唇苜蓿盲蝽未出现明显的寄主不连续分布现象,同时,气候屏障对黑唇苜蓿盲蝽存活率及有效种群数量影响也较小,推测这些可能是导致中国北方地区黑唇苜蓿盲蝽较小遗传分化的主要原因。
种群历史动态分析显示,当把所有种群作为一个整体进行分析时,相关参数均为负值,其中Fu and Li’sF*和Fu and Li’sD*为显著负值(表3),表明黑唇苜蓿盲蝽种群近期可能经历了扩张(Lietal., 2015)。错配分析结果同样证明,黑唇苜蓿盲蝽经历过种群的增长,发生过种群扩张(Cristianoetal., 2016)。寄主植物种类和数量是影响昆虫生命活动的主要因素,生态环境中高的寄主丰富度会增加昆虫的生存力和繁殖力使种群发生扩张。根据近年来棉花种植结构和面积的调整及棉盲蝽寄主广泛这一特点,可能出现由棉花经野生寄主或由棉花直接向其他作物扩散的情况,如Demirel(2009)在研究中发现曾经危害较轻的寄主作物油菜上苜蓿盲蝽种群密度显著增加密集的现象,类似的作物种群也可为黑唇苜蓿盲蝽提供了更多的食物选择和适宜的生存环境,作者推测寄主丰富度的增加是其发生种群扩张原因之一(Mengetal., 2018)。此外,黑苜蓿盲蝽的卵以棉花、马铃薯等农作物为载体,通过人为干扰或寄主植物携带的形式从而进行长距离扩散传播也是导致种群扩张的原因之一(李继强等, 2015; Caoetal., 2017)。
