二代测序技术检测家族性偏瘫型偏头痛患儿的基因突变
2020-03-02葛丽丽陈重芬刘磊郑璇宋银森
葛丽丽,陈重芬,刘磊,郑璇,宋银森
(郑州大学附属儿童医院&河南省儿童医院&郑州儿童医院河南省儿童遗传代谢性疾病重点实验室,郑州 450018)
家族性偏瘫型偏头痛(familial hemiplegic migraine,FHM)是一种罕见的常染色体显性遗传、具有先兆症状的偏头痛[1]。本病由Clark等于1910年首次报道,是目前唯一明确的单基因遗传偏头痛,临床主要表现为发作性单侧肢体无力,后出现偏头痛伴发恶心呕吐,其他先兆症状如视觉、感觉、言语等出现异常,通常在儿童或青春期起病[2]。本研究分析1例CACNA1A基因 c.4552G>A新生突变导致家族性偏瘫型偏头痛患儿的临床及遗传学特征,为完善和丰富该病的临床-基因表型提供实验依据。
1 材料与方法
1.1一般资料 患儿,男,3岁8月龄,因“抽搐、呕吐、左侧肢体无力1 d”于2018年8月收入我院神经内科。2岁8月龄时曾出现左侧肢体无力伴呕吐,1 d后自行缓解。今再次出现相似症状,并伴视物模糊,抽搐、头痛、独坐易向左侧倾倒。为求进一步治疗,收入我院。患儿系G1P1,足月顺产,生后无窒息抢救史,出生体重2.5 kg,身长不详。母孕期体健,家族中无遗传疾病史。FHM诊断标准参照HIS《国际头痛疾病分类》(2004年)第二版:头痛先兆必须包括完全可逆的肢体乏力,必须包括完全可逆的视觉、一般感觉和语言障碍这3项症状中的1项,家族中至少有一级或二级家属有同样的症状[3]。
1.2体格检查 神志清,精神、饮食欠佳,呼吸平稳,哭闹时口角右歪,左眼闭合不全,左侧鼻唇沟变浅。左侧肢体活动障碍,右侧肢体活动无受限,关节无红肿。双侧膝腱反射、跟腱反射可引出,左侧巴氏征阳性,左上肢肌力Ⅱ级,左下肢肌力Ⅲ级,肌张力低;右侧肢体未见明显异常。
1.3实验室检查 血常规、肝肾功能、电解质、心肌酶谱均正常;头颅脑磁共振成像:右侧颞顶枕叶及右侧海马脱髓鞘改变,右侧大脑半球萎缩,右侧脑室扩张。以上均由本院相关科室完成。
1.4基因检测 经患儿监护人签署知情同意书,并经医院医学伦理委员会批准。抽取患儿及父母空腹静脉全血各2 mL,EDTA-K2抗凝,送至北京迈基诺医学检验所,按照QIAamp DNA提取试剂盒(德国Qiagen公司)说明书抽提基因组DNA,对患儿外显子编码区进行测序,并通过PCR扩增以及Sanger测序技术对筛选出的位点进行验证。剩余全血及DNA样本置于-20 ℃保存。以同期本院100例体检健康者作为健康人对照。
1.5突变位点的生物信息学分析 利用SIFT、PolyPhen-2软件预测错义突变对蛋白质功能的影响,应用Mutation Taster软件评估突变位点在物种间的保守性。采用SMART网站(http://smart.embl.de/)预测该蛋白质结构域和功能分析。
2 结果
2.1基因检测结果 通过全外显子组基因测序发现,患儿CACNA1A基因第28号外显子存在c.4552G>A突变。Sanger测序验证显示,其父母该位点均为野生型,患儿携带c.4552G>A突变为自发突变(图1)。而在100例健康人对照者中均未检测到该突变位点,排除其为多态性位点。
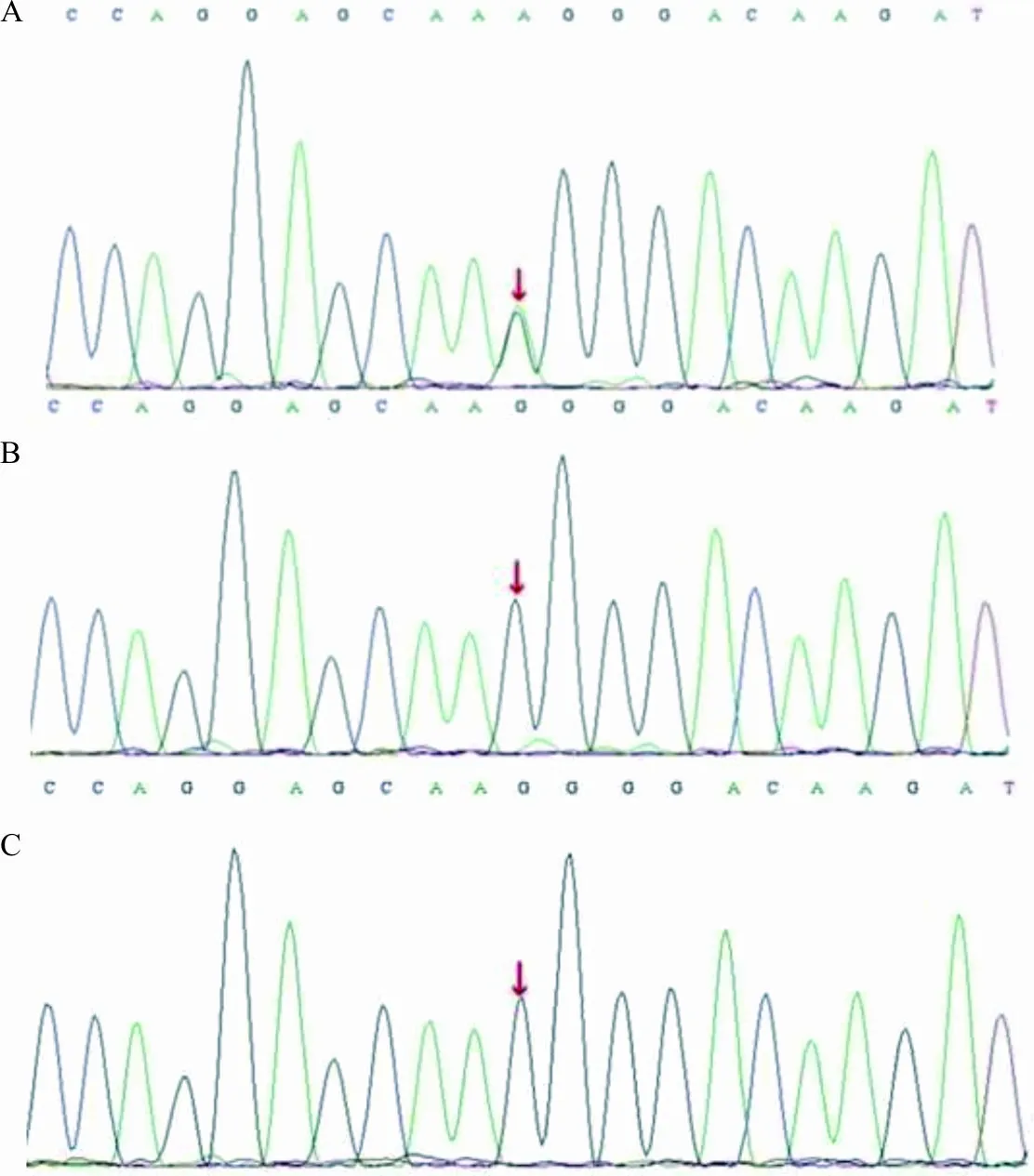
注:A,先证者携带c.4552G>A突变;B,父亲未携带c.4552G>A突变;C,母亲未携带c.4552G>A突变;箭头示突变位点。
2.2突变位点的生物信息学分析结果 c.4552G>A(p.G1518R)突变经蛋白质功能预测软件SIFT、PolyPhen-2预测结果均为“有害”(图2A、2B);Mutation Taster物种保守性分析显示,CACNA1A蛋白第1 518位氨基酸在不同的物种间几乎完全相同,表明在进化上高度保守(图2C)。根据SMART网站预测,该位点位于CACNA1A蛋白关键结构域。
3 讨论
FHM是一种罕见的常染色体显性遗传性疾病,是先兆偏头痛(migraine with aura,MA)中的一种亚型[4],多数在儿童或青春期起病,临床主要表现为偏头痛、可逆性单侧肢体无力及其他先兆症状如视觉、感觉、言语先兆等。李芳芳等[5]报道,一CACNA1A基因突变导致的家族性偏瘫型偏头痛家系中的患者均出现视物模糊,可逆性单侧肢体无力,持续10~20 min后自行缓解,随之出现对侧颞部疼痛,并伴恶心、呕吐、畏声、嗜睡。本例患儿出现视物模糊,10 min后出现左侧肢体无力,持续15 min后缓解,随后出现右侧颞部头痛、呕吐,并伴抽搐、哭闹时口角右歪,左眼闭合不全,左侧鼻唇沟变浅等临床症状。其临床表现与上述文献[4-5]报道的FHM的临床表现相一致。
本研究采用外显子组基因测序发现,患儿CACNA1A基因第28号外显子存在c.4552G>A突变,该突变导致第1 518位氨基酸由甘氨酸变异为精氨酸,为错义突变,经数据库检索,c.4552G>A突变未出现在EXAC、ESP6500及千人基因组计划数据库中,在100例健康人对照者中均未检测到该突变,排除其为多态性位点。Sanger测序验证显示,其父母该位点均为野生型,患儿携带c.4552G>A突变为自发突变。研究证实,50%~75%的FHM是由CACNA1A基因突变所致[6-7],已报道的CACNA1A基因突变形式有15种,且均为错义突变。本实验中检测到的CACNA1A基因p.G1518R突变为国内外尚未报道过的新突变,经蛋白质功能预测软件SIFT、PolyPhen-2预测结果均为“有害”。 根据SMART网站预测,CACNA1Ac.4552G>A突变导致第1 518位氨基酸由甘氨酸变异为精氨酸,该突变位点位于CACNA1A蛋白的关键结构域(ion trans domain)中,该结构域具有279个氨基酸,对应的氨基酸位点为1 243至1 521位,主要参与膜结构的形成、离子跨膜运输过程以及离子通道活性。根据Mutation Taster软件预测突变位点的致病性,得出结论:这一突变位点的改变,会引起蛋白质氨基酸序列改变,可能会影响该蛋白质的结构和功能。
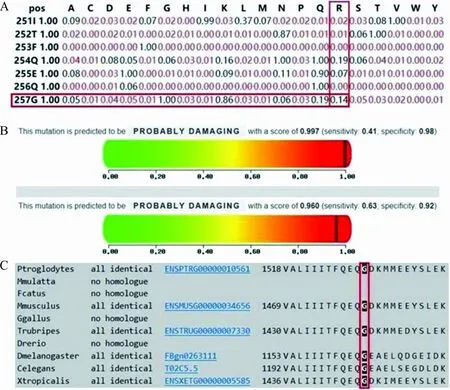
注:A,SIFT软件预测值为0.14;B,PolyPhen-2软件预测结果为0.997和0.960;C,Mutation Taster软件预测该位点在不同的物种间完全相同;各缩写含义:A,精氨酸;C,半胱氨酸;D,天冬氨酸;E,谷氨酸;F,苯丙氨酸;G,甘氨酸;H,组氨酸;I,异亮氨酸;K,赖氨酸;L,亮氨酸;M,蛋氨酸;N,天冬酰胺;P,脯氨酸;Q,谷氨酰胺;R,精氨酸;S,丝氨酸;T,苏氨酸;V,缬氨酸;W,色氨酸; Y,酪氨酸。
图2该突变位点的生物信息学分析
为进一步判定该位点的致病性,根据美国遗传学与基因组学学会(ACMG)发布的《ACMG遗传变异分类标准与指南》,该突变具备1个PS(突变位点为自发突变)+1个PM(在正常人群数据库中的频率为“-”,为低频),初步判定为“Likely pathogenic”。经蛋白质功能预测软件SIFT、PolyPhen-2预测结果均为“有害”,Mutation Taster物种保守性分析显示,CACNA1A蛋白第1 518位氨基酸在不同的物种间几乎完全相同,表明其在进化上高度保守。
FHM是一种遗传因素明显的MA,临床出现可逆性的单侧肢体无力,继而发生偏头痛,恶心呕吐等表现时,应早期进行基因检测,明确病因,及时治疗并控制发作。综上所述,本研究发现的CACNA1A基因c.4552G>A突变为国内首次报道的新突变,丰富了CACNA1A基因的突变谱,提高了临床医师对家族性偏瘫型偏头痛的认识,为FHM患儿的诊断和遗传咨询提供了重要线索。
