基于动态流变学的明胶/变性淀粉共混体系溶胶-凝胶相互转变动力学解析
2020-03-01邹秀容朱建华刘日斌
邹秀容,朱建华*,刘日斌,单 斌
(韶关学院英东食品科学与工程学院,广东 韶关 512005)
食品工业所囊括的各种产品中凝胶态食品占据重要的份额,食品凝胶通常是由生物高分子(蛋白、多糖)为结构材料形成的物理凝胶,其主要通过改变温度或调整离子强度、pH值等方法使生物高分子溶液体系完成溶胶-凝胶转变而获得。生物高分子体系溶液-凝胶相互转变过程非常复杂,受到高分子组分类别、浓度、离子强度、pH值以及共溶质类型和浓度的影响[1],部分食品物理凝胶加热时发生凝胶-溶胶相转变,由凝胶态可逆回归到溶胶态。
明胶软糖、琼脂软糖及卡拉胶软糖等凝胶糖果产品为典型的热可逆物理凝胶,此类凝胶糖果产品生产制备过程常以生物高分子(蛋白、多糖)为主要结构材料成分,在共溶质(蔗糖、葡萄糖浆等)场中通过溶胶-凝胶成型制备[2]。共溶质的分子质量虽远小于高分子组分,但可改变高分子组分在溶液中的分子折叠和缠结,并改变体系流变性质,进而影响到产品质构、风味释放等性质[3-4]。明胶软糖通常具备良好的弹性和耐咀嚼性等质构特性,广受消费者青睐。明胶软糖的主要成分明胶是由胶原质通过加热酸法或碱法水解获得,胶原质分子的天然构象主要由3 条多肽链形成的三螺旋结构构成并通过氢键稳定,多肽链上每3 个残基的重复序列为甘氨酸-脯氨酸-羟基脯氨酸(Gly-Pro-Hyp),水解之后获得的明胶分子在水中呈直链高分子状,具有无规卷曲构象[5]。
从材料学视角看,明胶软糖为一类天然高分子物理凝胶,其溶胶-凝胶制备过程原理主要涉及明胶分子发生线团-螺旋转变。明胶软糖制备过程单一使用明胶作为食品高分子基材时,其最终制品质构特性常存在弹性过大、咀嚼性及成本高等劣势,难以满足消费者和生产者需求,为此,食品工业生产过程通常采用明胶与变性淀粉复配来改善产品口感及降低成本[6]。复配过程实际上借鉴了材料科学研究领域的聚合物共混改性技术,以发挥明胶及变性淀粉各自的物性优点。但此类产品货架期容易出现析水、发烊等质量劣化问题,此类质量问题的突出特征为结构变形、崩解及液化而黏连外包装等,主要由制品发生凝胶-溶胶相转变导致结构解离引起。因此开展明胶/变性淀粉复合凝胶糖果溶胶-凝胶及凝胶-溶胶相转变过程特性研究具有重要的学术和应用价值。
小振幅动态流变测试因对受试材料自身结构不会造成破坏,且试样可呈现线性黏弹性质,对其形态结构响应性非常敏感,常被用于表征高分子材料的结构形态。近年来对明胶-多糖共混体系相关性质的研究引起关注[7],但迄今为止鲜见文献报道应用动态流变学研究共溶质场中明胶/多糖共混体系相转变动力学。本实验研究葡萄糖浆共溶质场中明胶/变性淀粉双组分模型共混体系溶胶-凝胶相互转变过程流变性质变化趋势,进而分析相转变动力学,以期为明胶软糖类凝胶制品工艺优化及质量控制提供应用理论指导。
1 材料与方法
1.1 材料与试剂
B型明胶(食品级、200 g冻力、pI 4.4~4.7、水分质量分数10.5%、灰分质量分数1.5%) 罗赛洛(广东)明胶有限公司;DM-803酸法变性淀粉(modified starch,MS)(水分质量分数≤14%、灰分质量分数≤0.5%,80 目筛通过率≥95%) 东莞东美食品有限公司;葡萄糖浆(glucose syrup,GS)(淀粉水解度42%) 广州双桥股份有限公司。
1.2 仪器与设备
SRT-202滚轴混合器 其林贝尔仪器制造有限公司;SHA-C恒温水浴振荡器 常州国华仪器有限公司;MCR92旋转流变仪 奥地利Anton Paar公司。
1.3 方法
1.3.1 明胶-变性淀粉共混溶液体系制备
12%(质量分数,下同)明胶储备液在80 ℃水浴中恒温30 min,以确保明胶颗粒全部水化溶胀分散。称取适量变性淀粉分散于纯水中,于90 ℃磁力搅拌水化5 min,配制均一的12%变性淀粉储备液。将明胶储备液和变性淀粉储备液等体积混合于烧杯中,在80 ℃下混合均匀并保温,获得6%明胶/6%变性淀粉储备液共混体系,随后加入相应葡萄糖浆混匀,配制含0%(对照)、20%、40%、60%及80%葡萄糖浆共溶质的明胶/变性淀粉共混体系。
1.3.2 动态流变学参数测定
含有不同质量分数葡萄糖浆的明胶-变性淀粉共混溶液于80 ℃恒温30 min后转移至保温桶中进行保温,然后将1 mL样品快速转移到预先加热到75 ℃的流变仪平行板(上、下板直径均为45 mm,狭缝宽度1 mm),除去过量样品并在样品外缘加一层轻质硅油防止样品中的水分蒸发。保持在线性黏弹区范围内的应变(γ)条件下(γ=0.5%)进行动态流变测试。
溶胶-凝胶转变过程测试程序:首先于75 ℃以2 ℃/min降温至5 ℃,然后在5 ℃恒温20 min,此过程角频率(ω)为6.28 rad/s,记录随时间(t)变化的弹性模量(G’)、损耗模量(G”)及损耗系数(tanδ);恒温末进行频率扫描,分析共混体系力学响应性质,频率扫描范围为0.1~100 rad/s,记录G’、G”及复数黏度(η*)随ω变化的情况。
凝胶-溶胶转变过程测试程序:前置降温恒温程序同上述溶胶-凝胶转变测试,然后从5 ℃以2 ℃/min升温至55 ℃,ω为6.28 rad/s,记录随t变化的G’、G”、tanδ;升温末进行频率扫描,频率扫描范围为0.1~100 rad/s,记录随ω变化的G’、G”及η*。
1.3.3 共混体系相转变过程结构形成及解离速率的计算
非等温或等温过程特定时间阶段共混体系凝胶或溶胶形成的相对快慢常用结构化平均形成速率(SDRa)、即时速率(v)进行表征[8-9]。SDRa可根据式(1)计算。

式中:G’ini及G’end分别为特定阶段对应的起始、终止G′/Pa;tini及tend分别为对应阶段起始、终止时间/s。
v可由式(2)计算。

式中:v为G’-t曲线求一阶导数得到的结构化即时速率/(Pa/s),v>0时为凝胶化速率,以vg表示;v<0时为溶胶化速率,以vs表示。
SDRa及v分别可用于表征时间段及时间点的结构化相对快慢,但尚难以定量表征结构化速率改变的快慢,为此引入结构化加速度(α),按式(3)计算。

式中:α为对G’-t曲线求二阶导数所得,记为即时加速度/(Pa/s2),加速度的正负性具有物理意义,其符号与v相同时表示结构化形成(或解离)处于加速阶段;其符号与v相反时视为结构化形成(或解离)处于减速阶段,用来辅助分析凝胶化或溶胶化加速过程特征。
1.3.4 溶胶-凝胶转变及凝胶-溶胶转变过程活化能的计算
降温过程非等温动力学模型常用于溶胶-凝胶转变动力学分析[10]。此种方法主要基于经典的非等温化学分解反应动力学方程[11](式(4))。
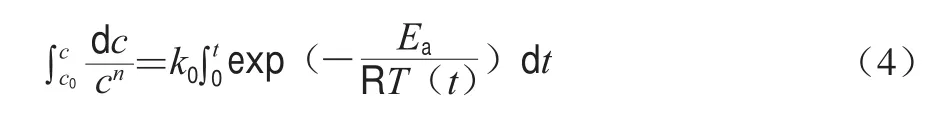
式中:c为反应物浓度/(mol/L);c0为反应物初始浓度/(mol/L);t为反应时间/s;k0为指前因子;Ea为反应活化能/(J/mol);T为t时刻的绝对温度/K;R为气体常数(8.314 J/(molgK)),此表达式依赖于变温速率。变温速率恒定的前提下,基于实验实证和回归分析推导获得式(5),其中Ea和k0可由Arrhenius关系图中计算,即通过对1/T作图确定。
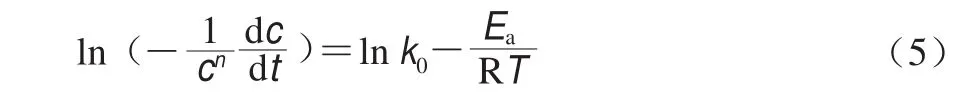
非等温条件下的经典化学反应动力学模型与流变测试黏弹性模量测试可借鉴橡胶弹性理论进行关联转换。具体就溶胶-凝胶相互转变体系而言,因G’通常与高分子交联结构数量或浓度成比例,因此浓度改变效应可通过同步改变的G’来研究,对方程(5),G’与c的改变具有等效性,且c和其变化值(dc)可进一步用G’及其变化值(dG’)予以替代[12],并得到直接用流变学参数表征相转变过程的动力学方程(式(6))。
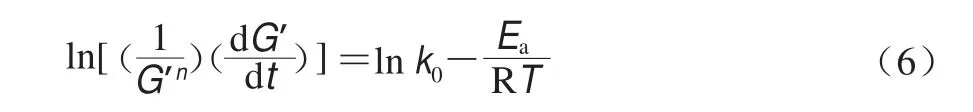
与公式(5)相比,溶胶-凝胶转变过程G′随时间延长呈增长趋势,此公式中负号转换为正号是因为凝胶形成过程G’增量为正,这与反应物浓度增量为负相反。生物大分子溶胶-凝胶过程通常取n=2[13-14]。物理凝胶的凝胶-溶胶转变过程因G’随时间延长呈递减趋势,此公式中仍保留负号是因为凝胶形成过程G’增量为负,与反应物浓度增量为负一致。
1.4 数据统计与分析
实验数据采用Origin 9.0软件作图,流变曲线及Ea求解拟合过程均采用最小二乘法。
2 结果与分析
2.1 葡萄糖浆共溶质质量分数对明胶/变性淀粉共混体系溶胶-凝胶转变过程流变性质的影响
2.1.1 溶胶-凝胶转变过程G’、G”随t的变化分析结果

图 1 明胶/变性淀粉共混体系溶胶-凝胶转变过程G’、G”随t变化曲线Fig. 1 G’ and G” versus t cures of GL/MS blends during sol-gel transition
由图1可知,不同质量分数葡萄糖浆共溶质场中明胶-变性淀粉共混体系降温阶段G’、G”-t曲线均为类“S”型。0~1 500 s阶段(约75~30 ℃)G’、G”相对较小,均低于10 Pa,共混体系仍然为可流动的浓溶液状态,明胶及变性淀粉分子主要处于无规卷曲状态。随温度下降,到特定温度点时,G’、G”从低模量基准线跃增。第二阶段随温度降低G’、G”均呈增加趋势,且差幅越来越小,G’=G”的温度点被定义为凝胶点(Tg)[15]。此温度点溶胶体系首次形成三维网络结构,主要因为共混体系中形成的三螺旋交联区域覆盖到整个溶液共混体系空间,共混体系不能再流动。随温度进一步降低直至接近降温程序末端(5 ℃),G’、G”进一步增加,且G’与G”增幅差逐渐加大,主要因为非凝胶区域逐渐有更多的明胶高分子形成三螺旋并加入和充密到既有三维网络空间所致[16]。结合表1可知,与未添加葡萄糖浆组相比,20%葡萄糖浆共溶质存在时共混体系G’1800s、G’降温末、G’恒温末由633.25、3 044.1 Pa及5 139.5 Pa分别降低为343.17、1 898.4 Pa及3 168.6 Pa,降低幅度分别达45.80%、37.64%及61.65%,且SDRa降温由1.41 Pa/s降低至0.88 Pa/s。随葡萄糖浆质量分数增加至40%~60%,G’1800s、G’降温末及G’恒温末进一步降低。80%葡萄糖浆共溶质存在时,与添加60%葡萄糖浆样品相比,G’降温末由1 074.3 Pa骤降至19.9 Pa,降低近两个数量级,表明添加80%葡萄糖浆共混体系形成的凝胶三维网络结构非常弱。
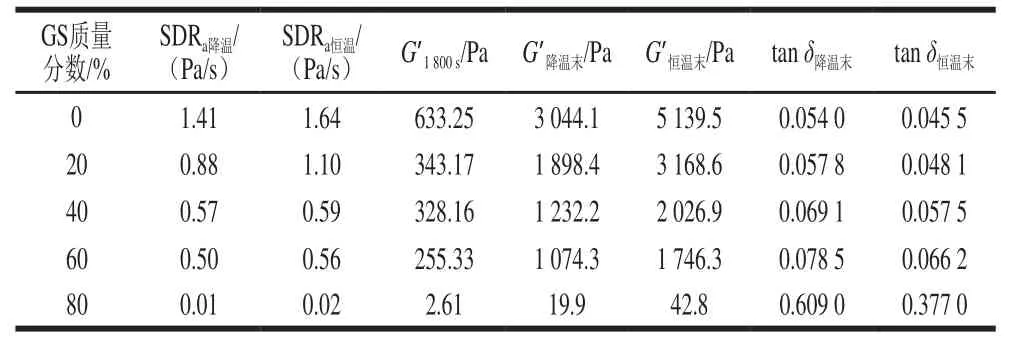
表 1 明胶/变性淀粉共混体系溶胶-凝胶转变过程流变学特征参数Table 1 Rheological characteristic parameters of GL/MS blends during sol-gel transition
2.1.2 溶胶-凝胶转变过程特征区域分析结果
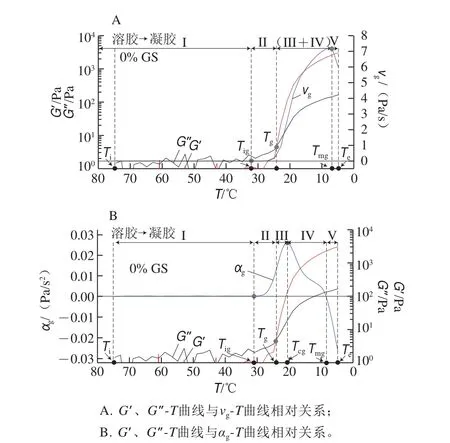
图 2 明胶/变性淀粉共混体系溶胶-凝胶转变特征温度解析图Fig. 2 Schematic illustration of the characteristic temperatures of GL/MS blends during sol-gel transition
G’、G”-T测定结果可较为清晰地定性分析明胶/变性淀粉共混体系溶胶-凝胶转变结构形成过程,将溶胶-凝胶起始阶段G’-T曲线分为诱导期、快速指数增长期及对数缓慢增长期,可以确定Tg点,但尚不能定量描述结构改变过程即时结构形成速率及其他关键温度点。为此对G’-T曲线求一阶导数和二阶导数,并转化得到相转变结构化速率曲线(v-T)和加速度曲线(α-T)。图2以未添加葡聚糖共混体系降温过程的vg-T、αg-T为例,对溶胶-凝胶转变过程进行分析。曲线vg-T或αg-T外推与vg=0或αg=0水平线(零线)相交的温度点定义为凝胶化起始点温度(Tig),此温度点确定方式示意图参照图3A中内置图;G’-T与G″-T曲线交点对应温度为凝胶形成点温度(Tg);vg-T曲线上vgmax对应温度定义为Tmg;αg-T曲线上αgmax对应温度定义为Tcg,此温度点不能直接从vg-T曲线上得出拐点对应温度,仅能从αg-T曲线上解析出。图2解析结果表明降温阶段除去降温程序起始温度点(Ti)和程序终点温度(Te),存在上述4 个特征温度Tig、Tg、Tcg及Tmg,溶胶-凝胶相转变过程5 个阶段的温度顺序为Tig>Tg>Tcg>Tmg。
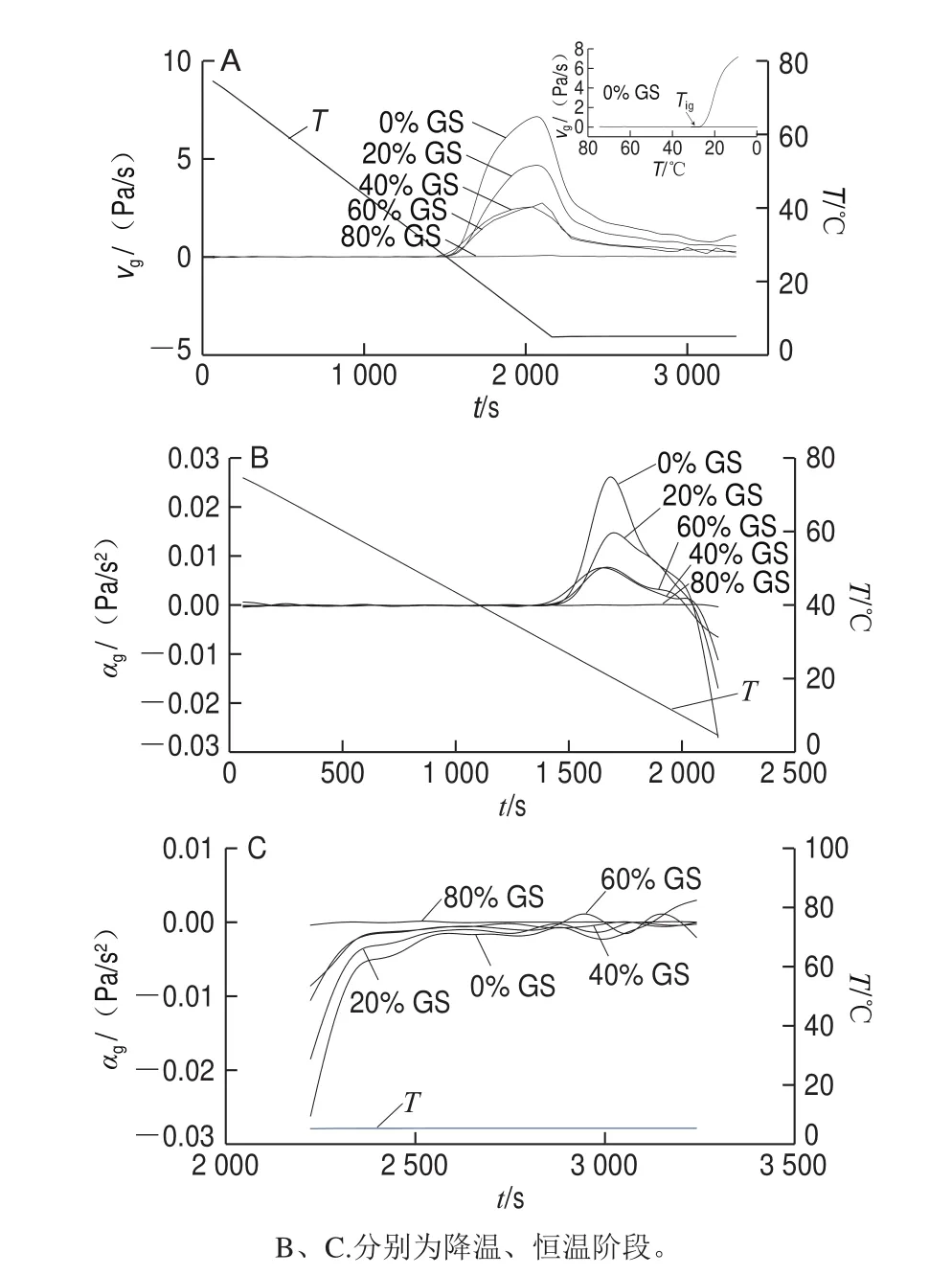
图 3 明胶/变性淀粉共混体系凝胶化速率(A)及加速度(B、C)随t变化曲线Fig. 3 vg versus t (A) and αg versus t (B, C) cures of GL/MS blends
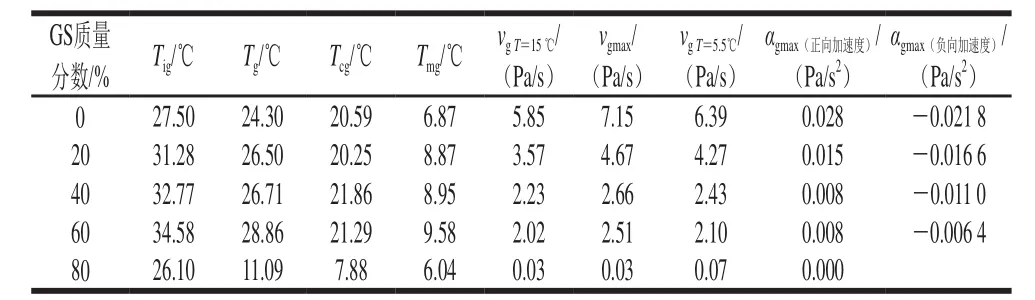
表 2 明胶/变性淀粉共混体系溶胶-凝胶转变过程特征温度及凝胶结构化参数Table 2 Characteristic temperatures and structure formation parameters of GL/MS blends during sol-gel transition
图3为降温恒温过程明胶/变性淀粉共混体系vg-t或αg-t曲线,表2为该过程结合图3解析得出的特征参数。与对照组相比,添加20%葡萄糖浆样品Tig由27.50 ℃提高至31.28 ℃,可能的原因是葡萄糖浆存在时凝胶结构化起始温度得到提高。随葡萄糖浆质量分数进一步增加至40%及60%,Tig进一步被提高,0%~60%葡萄糖浆质量分数范围Tg、Tcg、Tmg变化趋势与Tig相同,随共溶质质量分数增加,各特征温度均被提高,表明此质量分数范围葡萄糖浆中低分子糖类各成分存在大量的羟基与水分子形成较强氢键作用,打破了明胶高分子与水分子的相互作用,促进高分子由无规卷曲态向螺旋过渡态转变,并推动三螺旋聚集体形成,最终促成结构形成各特征温度提高。但80%葡萄糖浆存在时,与其他样品相比,显然共混体系凝胶结构形成被迟滞,Tig下降至26.10 ℃,Tg、Tcg、Tmg亦明显降低,共混体系凝胶SDRa急速降低。出现凝胶结构发展起始温度偏高但G’、SDRa、αg降低的可能原因主要是葡萄糖浆共溶质存在时明胶过渡态聚集物的长度减小,进而导致三螺旋聚集分子链缩短、三维网络结构弱化所致。不同体系小分子共溶质对高分子凝胶强度的影响作用及原因存在明显差异,Sharma等认为共溶质的存在阻断琼脂单股螺旋聚集,从而明显降低螺旋交联点的数量和密度,导致G’下降[17];Trombetta等通过跟踪水分活度发现多糖分子外围水化层水分子的减少限制了分子间螺旋的形成,并阻碍三维网络结构的形成[10];Evageliou等发现质量分数低于40%的葡萄糖浆对κ-卡拉胶凝胶网络结构可起到强化作用,主要因为其促进了卡拉胶分子无序-有序转变,但高于此值时G’下降[11]。
2.1.3 恒温阶段溶胶-凝胶转变流变性质分析结果
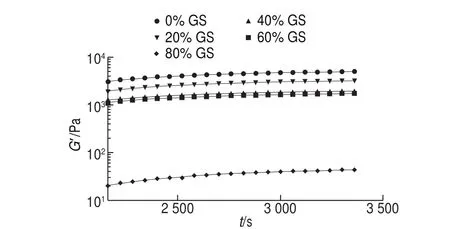
图 4 恒温阶段明胶/变性淀粉共混体系G’随t变化的拟合曲线Fig. 4 Fitted G’ versus t cures of GL/MS blends at constant temperature stage
图4为共混体系在恒温阶段中G’随t的变化曲线。结果表明虽然共混体系中含葡萄糖浆共溶质质量分数不同,但恒温过程中G’随t的变化规律非常相似。恒温阶段G’、G”增加进一步放缓,并趋近于平衡模量值。恒定温度条件下,交联区域的微区域调整重排及部分链段松弛,凝胶结构进入熟化和强化阶段,Goudoulas等研究明胶-海藻酸盐共混体系相转变动力学过程发现了类似的现象[18]。据图4中的曲线形状,G’-t关系符合式(7)关系。

式中:G’为恒温过程任意时刻的G’/Pa;G’∞为t=∞时的G’/Pa;m和k为大于零的常数,其中m反映凝胶结构达到G’∞时所需时间,而k为速率常数,反映G’随t变化速率的大小。

表 3 恒温阶段明胶/变性淀粉共混体系G’-t拟合曲线相关系数Table 3 Correlation coefficient of the G’-t fitted curves for GL/MS blends at constant temperature stage
用式(7)对图4中各曲线进行拟合并分析,得到相关系数结果见表3,随葡萄糖浆质量分数增加,G’∞及k逐渐明显减小,与降温过程速率变化的规律一致,说明葡萄糖浆质量分数越大,共混体系在恒温过程形成的凝胶结构越弱,且凝胶速率也明显降低。但是m随葡萄糖浆质量分数增加变化较小,不同共溶质质量分数共混体系转变至平衡凝胶结构的时间相当。等温阶段达到平衡结构的时间和凝胶强度无直接关联,主要是因为共溶质的存在削弱了凝胶强度,同时凝胶结构化速率也被阻滞。
2.1.4 恒温末共混体系频率扫描力学性质分析结果
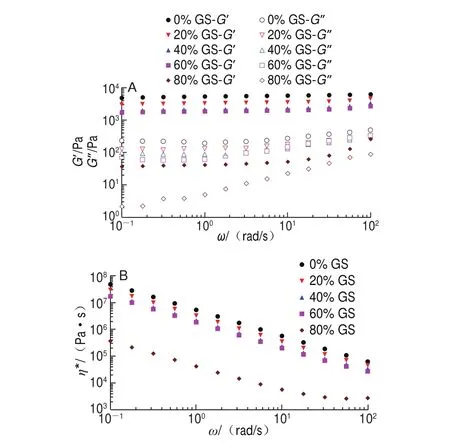
图 5 明胶/变性淀粉共混体系恒温程序末黏弹模量-ω(A)及η*-ω(B)曲线Fig. 5 G’ and G” versus ω (A) and η* versus ω (B) curves at the end of constant temperature stage
G’、G”及η*与ω的关系可用来反映受试材料的结构类型。恒温末频率扫描图(图5A)显示除添加80%葡萄糖浆共混体系样品在高频范围对频率依赖性较高外,其余样品全频率扫描范围G’、G”对ω的依赖均很小,且随葡萄糖浆质量分数增加G’逐渐降低。图5B显示对照组的η*均高于添加共溶质组,η*随ω的增加而减小,且整个频率范围0%~60%葡萄糖浆共混体系样品η*-ω双对数拟合直线斜率约为-1,表明均可形成较好的凝胶结构[19],但80%葡萄糖浆样品的η*在高频范围出现平台区,显示类似剪切变稀性质,表明凝胶结构非常弱。
2.2 葡萄糖浆共溶质质量分数对明胶/变性淀粉共混体系凝胶-溶胶转变过程流变性质的影响
2.2.1 凝胶-溶胶转变过程G’、G”随t的变化分析结果

图 6 明胶/变性淀粉共混体系凝胶-溶胶转变过程G’、G”随t变化曲线Fig. 6 G’ and G” versus t cures of GL/MS blends during gel-sol transition
图6显示,明胶/变性淀粉共混体系于5~55 ℃升温区间凝胶-溶胶相转变过程G’、G”随t变化,升温初期(5~18.5 ℃)G’、G”及两者差值开始呈减小趋势。随温度增加至18.5~30 ℃,G’、G”及两者差值进一步下降,但G′改变速率加大,直至G’=G”即为传统定义的溶胶点临界温度(Ts),此温度点维持临界三维网络结构,为体系可流动或形变的分界点。与溶胶-凝胶过程相比,添加相同葡萄糖浆质量分数共混体系的Ts均高于凝胶温度Tg,研究人员曾在明胶等热可逆凝胶体系中发现类似的溶胶点温度滞后于凝胶点温度的现象[20-21]。随温度进一步升高,G’、G”均降低且G’<G”,共混体系逐渐趋于溶胶化状态,最终明胶三螺旋结构不存在于体系中,明胶变性淀粉共混体系进入浓溶液状态,明胶分子的主要存在形式是无规卷曲。如表4所示,与对照组相比,添加20%葡萄糖浆的样品,G’升温始、G’t=300s、G’t=720s分别由5 651.9、4 489.9、386.5 Pa降低至3 643.2、2 934.8、225.8 Pa,平均溶胶化速率由3.77 Pa/s降低为2.43 Pa/s。80%葡萄糖浆共溶质存在时,样品的tanδ升温始明显大于添加0%~60%葡萄糖浆样品的对应值,表明80%葡萄糖浆样品升温过程中共混体系结构状态明显异于0%~60%样品,三维网络结构非常弱。

表 4 明胶/变性淀粉共混体系凝胶-溶胶转变过程流变学特征参数Table 4 Rheological characteristic parameters of GL/MS blends during gel-sol transition
2.2.2 凝胶-溶胶转变过程特征区域分析结果

图 7 明胶/变性淀粉共混体系凝胶-溶胶转变特征温度解析图Fig. 7 Schematic illustration of the characteristic temperatures of GL/MS blends during gel-sol transition
表 5 明胶/变性淀粉共混体系凝胶-溶胶转变过程特征温度及凝胶结构化参数Table 5 Characteristic temperatures and structure formation parameters of GL/MS blends during gel-sol transition
以未添加葡萄糖浆共溶质共混体系样品为例,通过对凝胶-溶胶过程G’-T曲线进行一阶和二阶求导得到溶胶化速率和加速度曲线,并获得另外4 个特征温度参数(图7):Tis为预溶胶起始关键温度点,即溶胶化速率达最大值时对应温度;Tcs为溶胶化加速度达最大值时对应温度点;Ts为溶胶态形成温度点,由G’-T、G”-T曲线交点确定;Tms为溶胶态完全转变点温度,由曲线vs-T或αs-T外推与vs=0或αs=0(零线)相交的温度点。这4 个特征温度值高低排序为Tis<Tcs<Ts<Tms,并将凝胶-溶胶化转变过程分为5 段。含不同质量分数葡萄糖浆共混体系特征温度见表5。
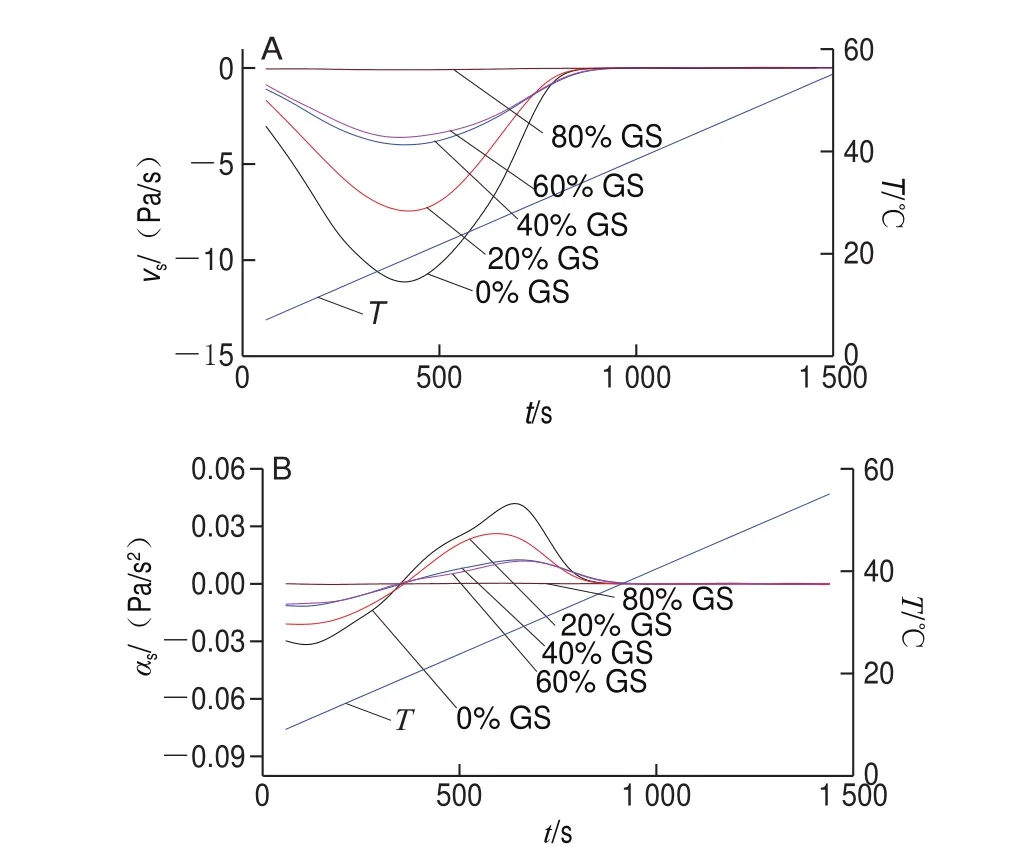
图 8 明胶/变性淀粉共混体系溶胶化速率(A)及加速度(B)随t变化曲线Fig. 8 vs versus t (A) and αs versus t (B) cures of GL/MS blends
图8及表5结果表明,随葡萄糖浆质量分数升高,溶胶化速率及溶胶化加速度均呈降低趋势。葡萄糖浆共溶质对Tis影响甚微,但明显降低了溶胶化速率,表明共溶质对预解离联结区形成没有影响,原因在于此过程仅涉及到构象熵调整,明胶分子链运动尚未开始。与对照组相比,添加20%葡萄糖浆后,明胶/变性淀粉共混体系的Ts由35.06 ℃降低至33.97 ℃,随共溶质质量分数增加,Tcs、Ts、Tms均呈降低趋势,其中对Ts影响最大,表明随葡萄糖浆质量分数增加,溶胶化进程被提前且呈加剧趋势,共溶质存在时利于凝胶结构发生解离。
2.2.3 升温末共混体系频率扫描力学性质分析
图9显示升温末G’、G”及η*随剪切速率增加呈现出两个具有明显差别的特征区域。当频率小于10 rad/s时,对照组及添加20%葡萄糖浆样品的流动性非常接近,但葡萄糖浆质量分数增加至40%、60%后,共混体系G’、G”对频率依赖性逐渐增强,η*下降明显,且提前到达平台区;葡萄糖浆质量分数增加至80%后,η*重新上升,可能由葡萄糖浆质量分数进一步增加后导致共混体系的黏度值升高而引起。当频率在10~100 rad/s范围时,受试样品的η*表现为随频率增加呈增加趋势,类似剪切变稠,内部结构重整。η*沿直线下降可以认为产生了剪切变稀溶液,归因于无规卷曲和缠结分子被打开,而低频率振荡过程因缠结未打开而不会出现剪切变稀现象[22]。
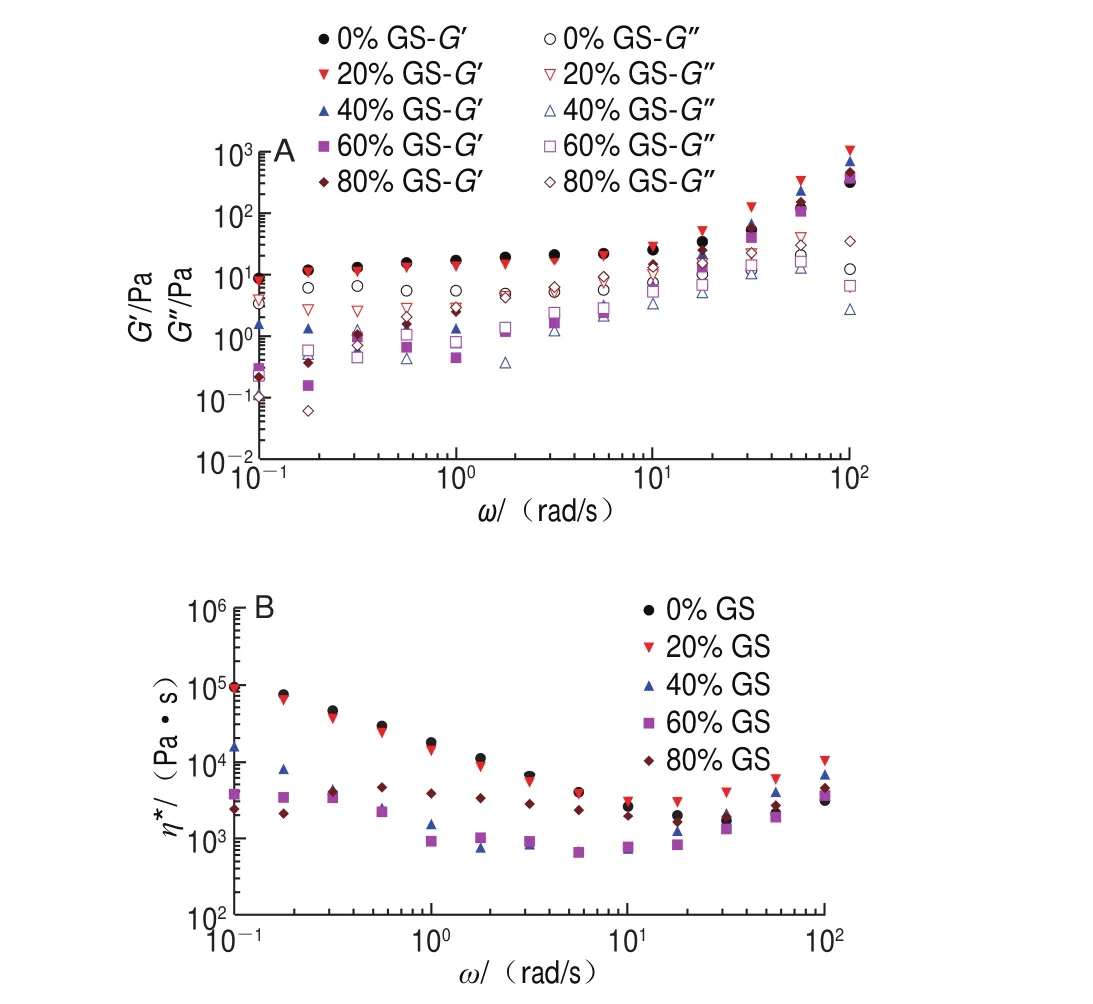
图 9 明胶/变性淀粉共混体系升温程序末黏弹模量-ω(A)及η*-ω(B)频率扫描曲线Fig. 9 G’ and G” versus ω (A) and η* versus ω (B) curves of GL/MS blends at the end of heating stage
2.3 葡萄糖浆共溶质场中共混体系非等温阶段相转变过程动力学分析结果
本研究采用的明胶/变性淀粉模型体系中,明胶组分直接对凝胶行为起作用,变性淀粉组分为低黏度酸化淀粉,主要利用变性淀粉占位效应使明胶于较低质量分数下形成凝胶。虽然明胶/变性淀粉共混体系流变学性质受到变性淀粉及葡萄糖浆的影响,但主要由明胶组分主导,图10为变性淀粉及葡萄糖浆分子拥挤环境下明胶溶胶-凝胶相互转变机理解析模式。

图 10 明胶/变性淀粉共混体系溶胶-凝胶及凝胶-溶胶转变机理示意图Fig. 10 Schematic presentation of the sol-gel and gel-sol transition of GL/MS blends
2.3.1 溶胶-凝胶相转变动力学解析
根据图10明胶/变性淀粉共混体系的相转变特征,提出简化后的溶胶-凝胶相转变动力学方程模型(式(8))。

式中:RC为无规卷曲;IH为过渡态螺旋结构;TH为三螺旋结构,即交联点;JZ为三螺旋交联区,即结区;NW为三维网络结构;n、α、β分别为有效结构单元数;k1~k4分别为各转变阶段的结构形成速率常数。
第I阶段对应温度区间为[Ti,Tig],共混体系处于高分子浓溶液状态,其中水为溶剂,葡萄糖浆为共溶质,明胶分子及变性淀粉分子呈无规卷曲状态,高分子链之间可接触、交叠,但尚无TH形成,体系可流动。明胶分子主要以RC及IH形式存在,且无规卷曲明胶分子浓度高于单螺旋或双螺旋过渡态分子浓度,即cRC>cIH;第II阶段对应温度区间为[Tig,Tg],对应高分子体系状态为交联态区(或称为预凝胶区),共混体系明胶高分子状态包括RC、IH、TH及JZ。明胶各种分子状态浓度排序为:cRC>cIH>cTH>cJZ>cNW,随温度下降,明胶三螺旋聚集体密度的增加,G’呈逐渐增加趋势[23]。第III阶段温度区间为[Tg,Tcg],对应高分子体系状态为凝胶前区,4 种结构状态均存在,随温度降低共混体系中cRC不断降低,单股螺旋增量(ΔcIH)逐渐升高,并在Tcg时达到最大值,cTH及cJZ逐渐升高,始终保持cIH>cTH>cJZ。第IV阶段为凝胶后区,温度区间为[Tcg,Tmg],ΔcIH逐渐降低,ΔcTH及ΔcJZ逐渐升高,但ΔcTH>ΔcJZ,直至ΔcTH达最大值,此刻凝胶化速度达vgmax。第V阶段温度区间为[Tmg,Te],既有明胶三螺旋聚集体不断转化为结区结构组分,ΔcTH不断降低,表现为vg降低,凝胶化网络不断强化,G’增加。研究人员提出明胶溶液体系发生溶胶-凝胶转变的机理存在较大差异,Djabourov等[24]提出明胶的凝胶过程主要涉及两个过程,首先随温度下降起始阶段明胶的三螺旋量以指数方式快速增加,第二阶段三螺旋量以对数方式增长。Bohidar等[25]提出明胶溶胶-凝胶转变过程存在3 个明显特征区域,首先是单体聚集,然后是无规-单股螺旋转变,紧接着发生单螺旋向三螺旋转变,并最终形成凝胶。
图11为未添加葡萄糖浆共混体系Ea求解过程拟合曲线示例,其余浓度葡萄糖浆共混体系Ea曲线趋势与此相同。由图11可知,溶胶-凝胶转变及凝胶-溶胶转变过程均在两个不同的温度范围(高温区及低温区)分为两个阶段反应。溶胶-凝胶转变及凝胶-溶胶转变过程高温区Ea均明显高于低温区,文献[9]发现类似现象。其中溶胶-凝胶转变过程对应解析图中的分解点温度为18.5 ℃(以对照组为例,与Tcg接近),高温阶段对应Ti~Tig~Tcg阶段,即对应解析图(图2)中I、II及III阶段,三螺旋过渡态形成阶段需克服更高Ea,此阶段k1、k2比较接近。而低温阶段对应IV及V阶段,即[Tcg,Te],转变快慢取决于明胶三螺旋聚集形成的形成速率,此阶段k3、k4比较接近。高温区Ea高于低温区,表明高温区明胶三螺旋形成量少的前提下,要形成网络需克服的能垒更高,低温区Ea低表明此阶段容易形成网络结构,因为低温区共混体系中易形成更多的三螺旋聚集物。
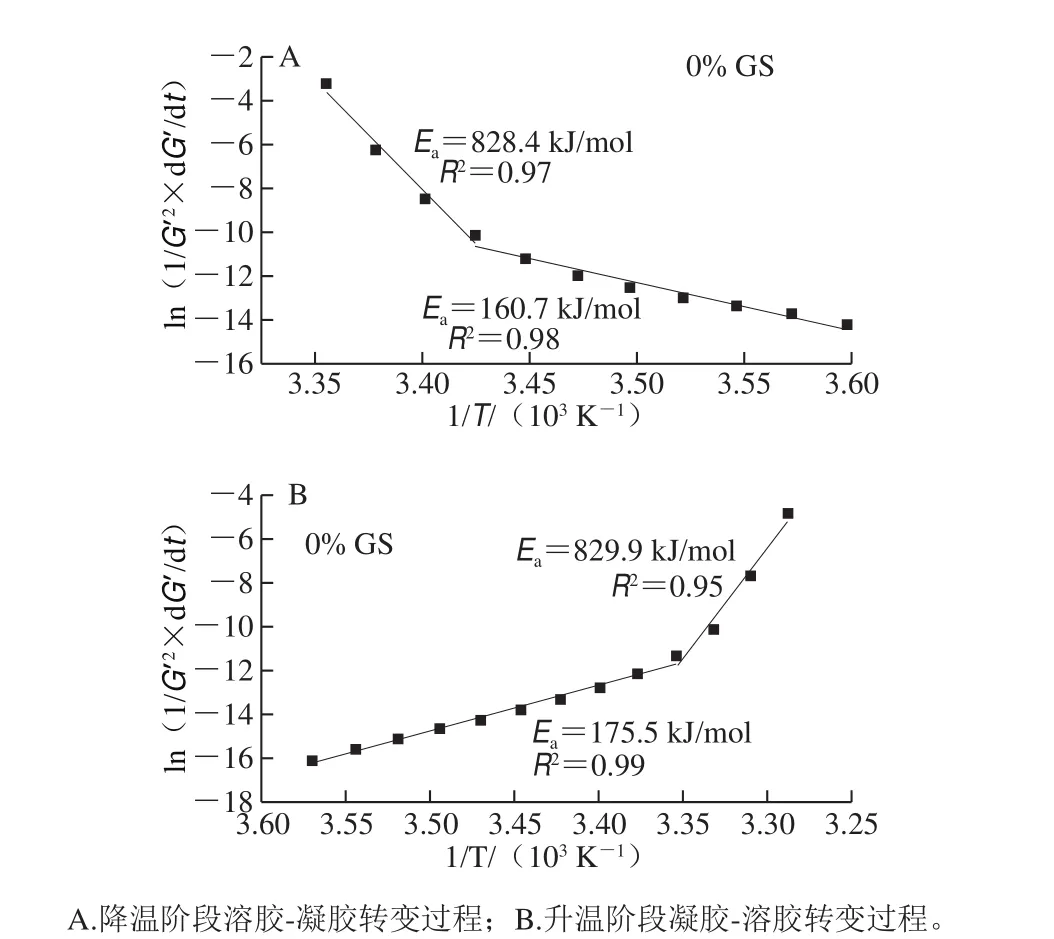
图 11 明胶/变性淀粉共混体系G’随温度变化的Arrhenius拟合曲线Fig. 11 Arrhenius fitting curves for G’ versus T cures of GL/MS blends
2.3.2 凝胶-溶胶相转变过程动力学解析
根据图10明胶/变性淀粉共混体系的相转变特征,凝胶-溶胶相转变动力学过程可简化为式(9)。
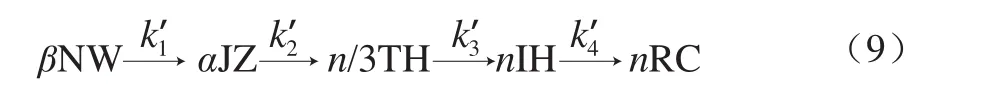
式中:k’1~k’4分别为各转变阶段的结构解体速率常数。
第I阶段温度区间为[Ti,Tis],体系为凝胶态,明胶分子以有序聚集TH形式存在于JZ中,后者构成三维网络结构,无规卷曲基本不存在。此过程溶胶化速率不断增加直至最大值(vsmax),伴随预解离JZ增量降低,但预解离联结区量增加,部分联结区解离形成预解离三螺旋,三螺旋含量逐渐相应增加。第II阶段温度区间为[Tis,Tcs],体系处于凝胶结构失稳前区,G’、vs继续下降,αs正向加速增加至最大值Tcs处,Tcs处三螺旋过渡态开始解螺旋。Nishinari等[26]提出“拉链模型”以解释热可逆凝胶-溶胶转变过程,他们认为当温度升至分子链段从拉链缔结区释放时,通过拉开分子拉链而触发凝胶-溶胶转变,与本研究结果类似。第III阶段温度区间为[Tcs,Ts],体系为凝胶态,温度驱动力减弱,处于凝胶结构失稳区,G’、vs均降低,αs正向加速降低,伴随对应微区联结区降低三螺旋量继续降低,单股螺旋量增加,自由卷曲链开始形成,直至微区联结区解离完成且三维网络结构骨架(晶种)解螺旋完毕,对应G’=G”,此后三维网状结构不复存在。拉链模型中提出凝胶-溶胶转变过程部分链段构型发生变化后,从局部区域开始逐渐扩散,只要有足够多的螺旋聚集存在就足以维持三维网络结构不致于崩塌[27]。第IV阶段温度区间为[Ts,Tms],体系为溶胶态,G’、vs均降低,αs正向加速继续降低,三螺旋含量继续降低,单螺旋增量逐渐减少,单股螺旋含量增加,自由卷曲链加速形成,自由卷曲链增速大于单股螺旋,且越来越大,形成剪刀差效应,直至临界点Tms三螺旋全部消失,体系中以自由卷曲和单螺旋存在。第V阶段温度区间为[Tms,Te],随温度进一步升高,共混体系高分子自由卷曲程度增大,高分子相互作用以缠结、互穿为主,呈浓溶液状态,流变学上体现为G”>G’,宏观和微区均不存在TH、IH及三螺旋过渡态。研究人员发现卡拉胶凝胶-溶胶转变过程存在类似现象[28]。
凝胶-溶胶转变过程对应解析图(图7)中的分解点温度为25.06 ℃(以对照组为例,与Tcs值接近)。低温阶段对应[Ti~Tcs]温度段(解析图中I及II阶段),受控于预解离三螺旋形成速率,只需克服更低的Ea,此阶段k’1、k’2比较接近。高温阶段对应[Tcs~Te]温度段(解析图中III、IV及V阶段),受控于三螺旋过渡态形成,需要克服更高能垒,此阶段k’3、k’4比较接近。Silva等[29]研究高甲氧基果胶/蔗糖复合体系时也发现高温区Ea明显高于低温区。共混体系凝胶动力学方程受试样的热处理历史影响明显,温度较高时Ea较大,主要因为缠结高分子与未缠结高分子之间存在较高的能垒,很难形成连续的网络结构,而温度较低区间的Ea较小,分子链之间容易发生聚集,并最终形成网络结构[30]。

表 6 明胶/变性淀粉共混体系溶胶-凝胶转变过程凝胶Ea及相关系数特征参数Table 6 Characteristic parameters of Ea and R2during sol-gel transition
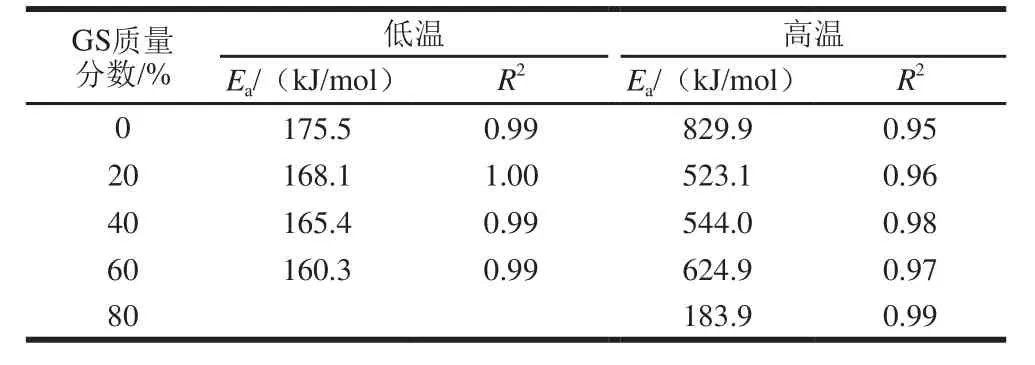
表 7 明胶/变性淀粉共混体系凝胶-溶胶转变过程溶胶Ea及相关系数特征参数Table 7 Characteristic parameters of Ea and R2during gel-sol transition
表6、7为降温及升温过程的Ea拟合结果,反映溶胶-凝胶及凝胶-溶胶转变过程。与对照组相比,20%葡萄糖浆共溶质存在时,高、低温区Ea分别由828.4、160.7kJ/mol降低至378.5、135.1kJ/mol,降低幅度分别达54.3%及15.9%,随葡萄糖浆质量分数增加,均出现先降低后增加趋势,但总体都低于对照样,可能因为葡萄糖浆共溶质存在而不利于网络结构形成且随共溶质质量分数增加而增加所致。葡萄糖浆质量分数对凝胶-溶胶转变低温区Ea影响较小。随葡萄糖浆质量分数增加,除添加80%葡萄糖浆样品外,其余样品高温区的Ea呈先降低后升高趋势,三螺旋解聚集阻力随葡萄糖浆共溶质质量分数增加而上升,但添加80%葡萄糖浆明胶/变性淀粉高温区Ea发生骤降,可能原因是体系中形成的明胶三螺旋聚集链短且数量少。
3 结 论
本研究提出一种依据流变学方法划分葡萄糖浆共溶质场中明胶/变性淀粉溶胶-凝胶转变及凝胶-溶胶转变过程的方法,溶凝转变及凝溶转变过程均存在4 个特征温度值,依据这4 个特征温度值可将相转变过程划分为5 个阶段,每个阶段具有明显的物理意义,其高分子存在及运动形式存在明显差别,流变性质及结构化速率具有明显差别。明胶/变性淀粉溶胶-凝胶转变及凝胶-溶胶转变过程G’、G”随葡萄糖浆共溶质质量分数增加明显降低,明显弱化了凝胶结构。随葡萄糖浆质量分数(0%~60%)增加,共混体系相转变过程凝胶点温度(Tg)呈上升趋势,而溶胶点温度(Ts)呈降低趋势,同时相转变过程及同温度点G’、相转变平均速率、即时速率及加速度呈降低趋势,表明葡萄糖浆共溶质的存在弱化了共混体系凝胶,且随葡萄糖浆质量分数增加而加剧。降温及升温相转变过程均存在两个Ea,高温阶段的Ea均高于低温阶段,随葡萄糖浆质量分数增加,共混体系相转变过程高温区Ea呈先降低后增加趋势,但低温区Ea不存在明显变化。
