人工RNA结合骨架的可溶性表达及其与寡核苷酸链的相互作用分析
2020-02-29徐如飞徐奕倩陈倩倩蒋培蓉陈美雯蒲首丞孙梅好
徐如飞,徐奕倩,陈倩倩,蒋培蓉,陈美雯,蒲首丞,孙梅好
(浙江师范大学化学与生命科学学院,浙江金华321004)
PUF结合因子(Pumilio/fem-3 binding factor)是一种存在于真核细胞、结合单链RNA分子的RNA结合蛋白,具有转录后调控蛋白质表达的重要功能。PUF由RNA结合骨架(RNA-binding scaffold,RBS)和辅助结构域构成,其RNA结合骨架大部分含有由α-螺旋构成的8个RNA结合域(RNA binding domain,RBD),天然PUF碱基特异性的解析为人工设计特异性结合任意8个核苷酸RNA分子的RBS(engineered RBS,ERBS)奠定了基础[5]。
PUF蛋白家族是RBP中的一个大家族,存在于所有真核生物中,其基因的拷贝数在不同物种中差异很大。PUF蛋白主要通过特异性结合目标mRNA的顺式作用因子,影响mRNA的稳定性、定位和翻译过程,参与胚胎形成、细胞发育和分化等过程,实现基因的转录后调控[6-8]。
据报道,在原核细胞中,PUF蛋白结合于靶基因的RBS和起始密码子之间,调控翻译;在真核细胞中,PUF蛋白结合于靶mRNA的5′非翻译区,显著抑制了靶基因的表达[9]。调控机制可能是由于PUF蛋白结合RNA之后,与其他的蛋白相互作用,进而抑制翻译或引起mRNA降解[10]。
利用Escherichia coli等原核表达体系表达重组蛋白,由于表达水平高及二硫键发生错配,导致80%会以不溶性的包涵体形式存在[11-12],在分离纯化蛋白之前,需要经过变性和复性,变性的蛋白质因为构象被破坏,不具备生物学活性。因此,需要通过复性使蛋白质恢复其天然构象状态,具有正确的生物学功能。变性和复性的过程比较复杂,而且难度大,因此,可溶性表达重组蛋白具有很重要的生物学意义[13-14]。
据报道,提高重组蛋白的可溶性表达可以采用以下几种方式:第一,选择合适的宿主,BL21(DE3)是通常选择的宿主细胞[15];第二,构建蛋白,比如将目的蛋白与一些可溶性蛋白表达,可以提高目的蛋白的溶解性,但是分离纯化之后需要切除辅助蛋白[16-19];第三,降低培养温度,大肠杆菌的最适生长温度为37℃左右,因此,37℃诱导蛋白很容易导致不溶性的包涵体形成,研究显示,低温诱导更有利于正确蛋白的折叠,导致其可溶性增加[20-24];第四,改善诱导条件,在菌体生长的对数生长期诱导,更有利于可溶性蛋白的形成,降低诱导剂的使用量有利于蛋白的缓慢表达,从而更有利于可溶性蛋白的形成[25]。
本研究利用ERBS蛋白可以结合单链RNA的特点,设计5'-FAM标记寡核苷酸,通过检测5′-FAM标记寡核苷酸荧光强度的变化来分析ERBS蛋白与寡核苷酸的相互作用。
1 材料和方法
1.1 材料
Escherichia coliDH5α、BL21(DE3)、Rosetta(DE3)感受态细胞、pET-24a-CYFP载体均由浙江师范大学化学与生命科学学院细胞实验室保存;含ERBS基因的载体pUC57-ERBS、寡核苷酸链由生工生物工程(上海)股份有限公司合成;限制性核酸内切酶Not I、Sal I、PCR相关试剂、T4 DNA Ligase等购自宝生工程(大连)有限公司;IPTG、氨苄青霉素、卡纳霉素、酵母提取物、胰蛋白胨均购自Oxoid LTD公司(Hampshire,England);DTT、溶菌酶、Pepstatin A购自Amresco公司;PMSF为BBI产品;Ni-NTA His-Bind SuperflowResin购自Novagen公司;超滤管购自Millipore公司。其他常规生化试剂均购自国内公司,为分析纯。
1.2 构建载体所需引物
根据已知的基因序列,用Primer premier 6.0设计引物,并由生工生物工程(上海)股份有限公司合成,所用引物如表1所示。通过引物ERBS-F与ERBS-R进行PCR扩增,得到含有Not I、Sal I酶切位点的ERBS,从而将其构建到pET-24a-CYFP载体中。
3.1.2 清创 采用自溶性清创法进行清创[6-7]。严格执行无菌技术,以0.9%氯化钠棉球彻底清洗伤口,将坏死组织及脓性分泌物清除干净。水凝胶均匀涂抹于坏死区,无粘性泡沫敷料保湿覆盖,低敏宽胶带封贴[8]。第2天就诊时发现渗液中等量,黑色坏死组织变软并且部分脱落,按照保守性锐器清创指南使用无菌剪刀,用血管钳分离和提起坏死组织并剪除[5]。
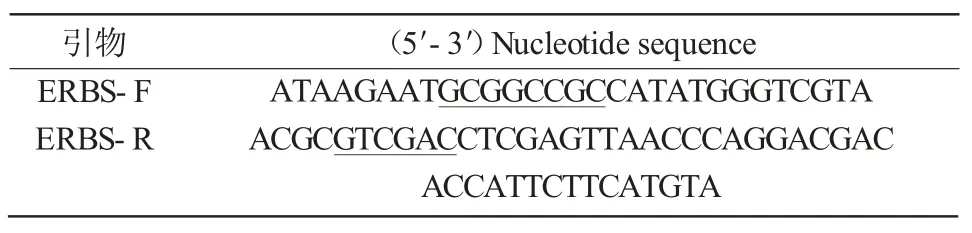
表1 构建载体所用引物
1.3 合成寡核苷酸链
设计ERBS特异性识别并结合的寡核苷酸,5′加FAM荧光基团(表2)。

表2 合成的寡核苷酸链
1.4 方法
1.4.1 pET-24a-ERBS表达载体的构建设计可以结合5′-ACAGCGGC-3′的ERBS基因序列,合成于pUC57-SmalI;以pUC57-ERBS为模板,利用引物ERBS-F和ERBS-R扩增之后,其PCR纯化产物与pET-24a-CYFP质粒通过双酶切(Not I、Sal I)及割胶回收后,二者的割胶回收产物经16℃连接过夜,将连接产物转化至DH5α,酶切验证正确之后转化BL21(DE3),获得pET-24a-ERBS表达菌株。
1.4.2 BL21(DE3)中ERBS蛋白的表达挑取pET-24a-ERBS表达菌株单菌落至含4 mL LB培养基(含Kana抗性)的试管中,37℃220 r/min摇床振荡培养过夜。将试管菌液转移至500 mL LB培养基中扩大培养,培养至OD600为0.4~0.6时,加入IPTG至终浓度为0.1 mmol/L,16℃诱导过夜后,离心收集菌体,使用磷酸钠溶液(pH=8.0),加入pepstatinA、PMSF、溶菌酶,4℃裂解1 h,超声波破碎后离心,分离上清及沉淀,将样品处理后进行SDS聚丙烯酰胺凝胶电泳。
1.4.3 Rosetta(DE3)中ERBS蛋白的表达提取pET-24a-ERBS(BL21)质粒,转化Rosetta(DE3)。挑取单菌落至含4 mL LB培养基(含Kana抗性)的试管中,37℃220 r/min摇床振荡培养过夜。将试管菌液转移至500 mL LB培养基中扩大培养,培养至OD600为0.4~0.6时,加入IPTG至终浓度为0.1 mmol/L,16℃诱导过夜后,离心收集菌体,使用磷酸钠溶液(pH=8.0),加入pepstatinA、PMSF、溶菌酶,4℃裂解1 h,超声波破碎后离心,分离上清及沉淀,将样品处理后进行SDS聚丙烯酰胺凝胶电泳。
将破碎上清通过镍柱纯化,采用0、5、50、100 mmol/L咪唑梯度洗脱目的蛋白,蛋白电泳显示,100 mmol/L咪唑可以将目的蛋白完全洗脱下来,将100 mmol/L咪唑洗脱溶液进行透析获得的蛋白样品,利用3 ku超滤管低速离心浓缩蛋白。蛋白浓度根据Bradford法[20]测定。
1.4.4 光谱分析利用安捷伦Cary 4000紫外-可见-近红外分光光度计的Kinetic程序,在激发波长为494 nm、发射波长为521 nm的条件下,测定不同浓度的ERBS蛋白与5′-FAM标记寡核苷酸相互作用的吸光值,计算解离常数。
2 结果与分析
2.1 ERBS的基因序列(5'-3')及其对应的氨基酸序列
以人的PUM1的RBD(G828-Y1168,PDB:1M8Y)为骨架,根据已经解析的RBD对应RNA的4种碱基的识别密码,设计ERBS的氨基酸序列,送至生物工程公司优化、合成大肠杆菌表达ERBS蛋白所需的核苷酸序列(图1),黄色区域为ERBS的8个RNA结合结构域,3′-5′依次结合RNA碱 基ACAGCGGC。
2.2 ERBS片段的获得
ERBS片段使用ERBS-F、ERBS-R引物通过PCR扩增得到,其扩增结果如图2所示,产物长度为1 000 bp左右,符合预期。割胶回收后酶切(Not I+Sal I),然后连接到pET-24a-CEYFP载体上,通过菌液PCR筛选出阳性克隆,构建重组质粒载体pET-24a-CEYFP-ERBS。
2.3 蛋白纯化结果
2.3.1 BL21(DE3)中ERBS蛋白的表达结果为了分析ERBS与其对应核苷酸的特异性互作,将ERBS通过BL21(DE3)表达菌株进行表达,并收集其破碎上清与沉淀进行SDS-PAGE电泳,结果显示(图3),蛋白诱导成功但大多数蛋白都形成了包涵体存在于沉淀中,包涵体的纯化需要经过复杂的变性复性过程,给后续的试验造成了很大的麻烦。
2.3.2 Rosetta(DE3)中ERBS蛋白的表达结果为了获得可溶性的ERBS,提取pET-24a-ERBS(BL21)质粒,转化Rosetta(DE3)进行表达纯化,SDS-PAGE电泳结果显示(图4),蛋白诱导成功,且为可溶性蛋白,并且通过镍柱纯化之后得到了纯度较高的ERBS(≥95%)。
2.4 ERBS蛋白与寡核苷酸链的相互作用
为了分析ERBS与其对应核苷酸的特异性互作,利用镍柱纯化的ERBS和5′-FAM标记寡核苷酸(0.4μmol/L)结合ERBS后荧光强度发生改变这一特点,分析了ERBS与其目标寡核苷酸(底物)之间的解离常数(Kd)。利用0.4μmol/L的5′-FAM标记寡核苷酸链,分别加入不同浓度的ERBS(图5),激发波长为494 nm,发射波长为521 nm,根据寡核苷酸链荧光强度的变化拟合获得解离常数Kd。数据拟合分析发现,Kd为(9.76±0.62)nmol/L,与已发表数据(Kd≈0.5~30 nmol/L)一致。
3 结论与讨论
设计可以结合任意RNA序列的PUF蛋白具有许多潜在的生物和医学应用[26]。RNA复合体在调节基因表达方面具有很重要的作用,对多细胞真核生物的细胞分化和复杂的发育模式也是至关重要的[27-29]。但是蛋白质以何种方式结合RNA很难预测[30],这限制了为生物技术和医学应用设计RNA结合蛋白[31]。RBP的潜能与siRNA和miRNA相似,然而,miRNA主要是降低靶RNA在细胞质中的丰富度或表达,这依赖于RNA干扰途径[32]。因此,设计RNA结合蛋白很吸引人,因为它们可以与任意感兴趣的结构域融合,选择性地结合于特定的RNA靶位点,进而监测或控制其代谢的任何方面。
人工设计PUF(Engineered PUF,EPUF)可以与任何靶RNA的几个碱基特异性地结合,招募其他蛋白因子形成功能性融合蛋白,从而调控mRNA剪切,影响蛋白质翻译和特定mRNA的稳定性。这些结果暗示,EPUF可以作为潜在的生物化学研究和生物医疗工具进行开发。
本试验为了分析ERBS与其对应核苷酸的特异性互作,通过Escherichia coli Rosetta(DE3)表达ERBS,并利用镍柱纯化获得ERBS,利用5′-FAM标记寡核苷酸(0.4μmol/L)结合ERBS后荧光强度发生改变这一特点,分别加入不同浓度的ERBS(激发波长为494 nm,发射波长为521 nm),根据5′-FAM标记寡核苷酸荧光强度的变化拟合获得解离常数。
Rosetta(DE3)补充6种稀有密码子(AUA、AGG、AGA、CUA、CCC、GGA),提高外源基因,尤其是真核基因在原核系统中的表达水平[33]。试验也证明,使用Rosetta(DE3)之后,确实提高了蛋白的可溶性表达水平,有关如何提高外源基因可溶性表达的策略,仍需要进一步探索。
