钙钛矿结构ZrBeO3稳定性的第一性原理研究
2020-02-25温新竹彭玉颜刘明真
温新竹,彭玉颜,刘明真
(仰恩大学工程技术学院,福建 泉州 362014)
钙钛矿氧化物(ABO3)由于具有铁电、铁磁、超导、压电等特性,在光波导、激光倍频、压电传感器、可调谐电容器、高容量存储单元等方面具有广泛的应用[1-4]。Park等[5]发现在电荷有序的短周期LaVO3/SrVO3钙钛矿超晶格中,通过调控钒离子价态可以形成拥有很大极化强度的亚稳态。Fennie[6]在寻求强磁电耦合材料的过程中发现PbTiO3/BiFeO3电磁耦合机制不同。Sonali等[3]计算了顺电BaTiO3的电子结构、化学键、光学性质,揭示出其应用的巨大潜力。常见锆酸盐类物质(AZrO3,A=Ca,Sr,Ba等)一般具有高熔点、高热膨胀系数、低热导率、高化学稳定性、高辐射稳定性,以及优良的离子导电性能,在热障涂层材料[7]、核材料[8]、耐火材料[9]、发光材料[10-12]、高温离子传感器件[13-14]等诸多领域得到广泛应用。目前关于ABO3结构与性能方面的计算预测研究众多,相对于锆基功能材料的广泛应用,ABO3型结构的锆酸盐晶体材料的理论研究较少[15-16],而对于Zr元素与B、O可能构成的两种ZrBeO3、BeZrO3结构几乎没有任何相关报道。本研究基于密度泛函理论(Density functional theory,DFT)的第一性原理,采用模守恒赝势平面波方法,通过对ZrBeO3的化学键Mulliken布居值以及声子谱、声子态密度的计算分析,从电子结构角度出发探讨ZrBeO3稳定性差的本质原因,研究结果对于进一步优化ZrBeO3的稳定性,缩短其实验研究及应用进程,以及开发Zr基多功能材料提供一定的理论参考。
1 理论模型与计算方法
1.1 理论模型建立
ABO3型钙钛矿的结构为简立方晶格,空间群晶胞中有5个原子,B占据立方晶格体心位置,A占据8个顶点位置,晶胞6个面心位置由O离子占据,O离子与中心B位离子形成BO6八面体。整个晶体也可看成由BO6八面体共顶点连接而成,各八面体之间的空隙位置由A位离子占据,A、B离子与O离子的配位数分别为12和6,一般而言,若要组成ABO3型钙钛矿结构,A、B、O的离子半径应满足紧密堆积原则

式中:RA、RB和RO分别为A、B和O离子的半径;t为容忍因子,一般情况下,t介于0.78~1.05[17]之间时,可组成钙钛矿结构,当t=1时,体系为理想钙钛矿结构。
由于Be2+、Zr4+、O2-离子半径分别为 0.17、0.72、1.21Å[18-19],若要满足空间几何结构稳定,根据式(1),几种元素只能组合成Zr占据晶胞顶点位置的ZrBeO3钙钛矿结构,其容忍因子为0.989,非常接近理想钙钛矿结构。经过多次模拟和优化,最终建立的ZrBeO3晶胞的晶格常数为0.346 1 nm。
1.2 计算方法
计算在Materials studio 2017软件的CASTEP[20]模块中完成。在密度泛函理论框架下[21],采用广义梯度近似(General gradient approximate,GGA)方法[22-23],其中交换关联函数选择Perdew-Burke-Ernzerhof(PBE)函数[24],几何结构优化采用 Broyden-Fletcher-Goldfarb-Shanno (BFGS)算法[25-26],声子能谱计算采用线性响应方法,布里渊区的积分采用以G点为中心6×6×6的K点设置。考虑过渡金属元素Zr的计算精确性,计算中离子实与价电子之间的相互作用选用模守恒赝势[27]描述,具体的电子组态分别为 Be(1s22s2)、Zr(4s24p64d25s2)、O(2s22p4),平面波截止能Ecut为 898.0 eV。结构的自洽优化收敛标准设置为:最大位移5.0×10-4nm,最大内应力收敛标准0.02 GPa,原子间的最大相互作用力0.01 eV/nm,结构的总体能量收敛小于5×10-6eV/atom。
2 计算结果及分析
2.1 晶体结构模型及热力学稳定性
至目前为止还没有形成稳定的ZrBeO3晶体化合物,因此未检索到相关实验数据,在理论计算方面Materials project平台数据库中有所有元素经过高通量计算的初步结构筛选数据,本研究在了解该平台晶体结构信息的基础上建立晶体模型,明确Be、Zr、O元素在晶胞中各自占位后,去除晶胞的所有对称性,对晶胞常数a、b、c在0.138~0.432 nm(晶胞中原子接近重叠至晶胞中各原子不能成键,取值间隔0.01 nm)大范围内分别取任意值进行结构充分弛豫,发现弛豫后结果均为晶格常数在0.34 nm附近的立方钙钛矿结构,说明钙钛矿构型是该三元化合物中最稳定相。进一步以a=0.34 nm为中心作钙钛矿构型ZrBeO3的晶格常数a与总能的变化曲线,如图1所示,可得出最低能量点-2 627.83 eV处于晶格常数a=0.347 nm附近,对此晶格常数建立模型,再次优化后得到最终模型晶格常数为0.346 1 nm的钙钛矿结构,如图2所示,晶胞相关参数列于表1。
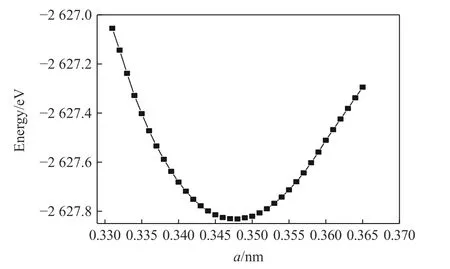
图1 ZrBeO3晶格常数a随总能E变化曲线Fig.1 Lattice constants (a) vstotal energy (E) of ZrBeO3
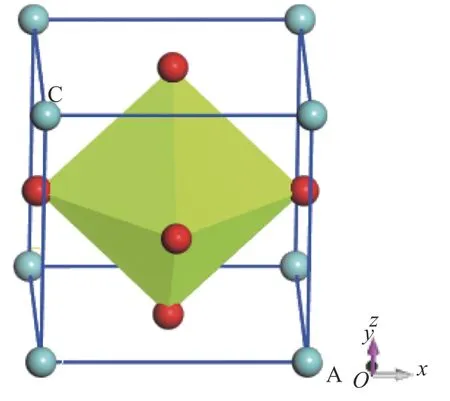
图2 ZrBeO3晶体模型Fig.2 ZrBeO3 crystal model
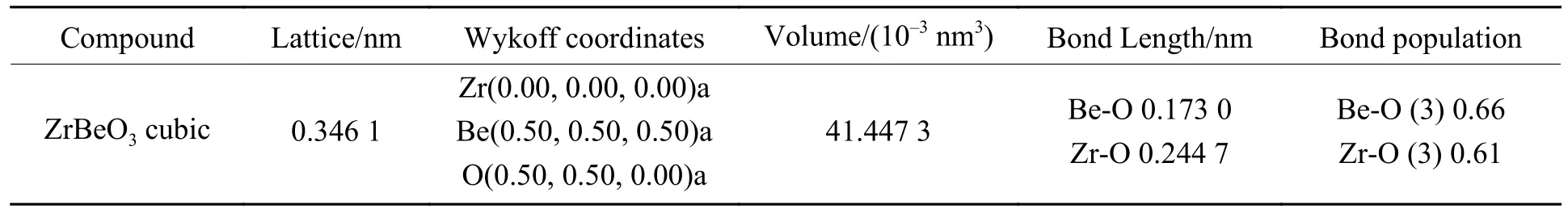
表1 ZrBeO3模型晶格参数Table 1 Lattice parameters of ZrBeO3 model
定义ZrBeO3结合能[28]为Ef=Et-EZr-EBe- 3EO,其中Et为结构充分优化后的钙钛矿型ZrBeO3的总能量(-2 627.83 eV),EZr、EBe、EO分别为 Zr、Be、O 的单自由原子能量,Ef为-38.27eV(-7.65 eV/atom),说明ZrBeO3钙钛矿构型具有较强的热力学稳定性。
2.2 ZrBeO3晶体结构的弹性常数及机械稳定性
立方晶系独立弹性常数C11、C12、C44描述了晶体对外加应变的响应刚度,计算了钙钛矿结构ZrBeO3在0~300 GPa不同压力下的弹性常数,如表2所示。结合玻恩-黄稳定性判据[29-30]

进行判断,所计算的ZrBeO3在不同压力下的钙钛矿结构在力学方面均满足稳定性要求。
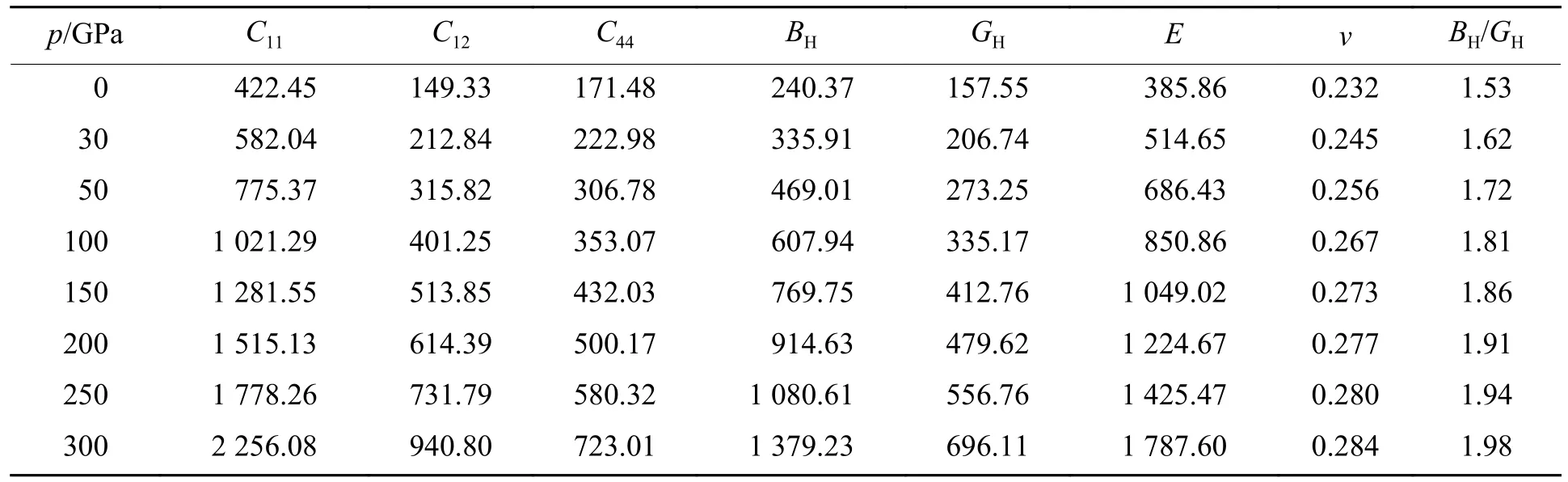
表2 不同压力下的弹性常数、体积模量、剪切模量、杨氏模量、泊松比、BH/GHTable 2 Elastic constants,bulk modulus,shear modulus,Young’s modulus,Poisson’s ratio,BH/GH under different pressures
根据Voigt-Reuss-Hill近似,立方晶系体模量BH、剪切模量GH、杨氏模量E、泊松比v可以根据式(3)~式(6)计算[31],结果如表2所示。
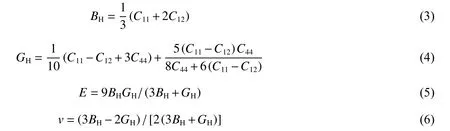
从表2中数据可以看出,ZrBeO3体弹性模量、剪切模量都比较大,理论上物质的硬度与体弹性模型密切相关,按一般经验理论初步判断它可能具有超硬性。Gao[32]基于第一性原理提出一种基于共价轨道重叠布居数的固体本征硬度值计算方法


式中:A为常量,取740,上标Be-O/Zr-O代表其化学键;P为布居值,Pμ为μ类型化学键的Mulliken重叠布居数; υb为 键体积(νb=V/n;V为原胞体积,n为原胞中所含该类键的数量);为μ类型化学键的体积;上标μ、v表示原胞中不同价键成分;dμ为键长;为单位体积内μ键的个数;Ω为晶胞体积;Nμ为晶胞内μ键的总数。代入表2中数据,计算得到钙钛矿结构的ZrBeO3常压下硬度约为34.5 GPa,超过SiC的硬度[32],在超硬应用材料方面应该有较大开发潜力。材料泊松比v为固体材料最大拉伸强度与最大剪切强度之比,表征材料受挤压或拉伸时的收缩率或膨胀率[33],按照断裂行为的判据,低泊松比材料属于脆性材料,这一点在BH/GH得到验证:通常认为BH/GH> 1.75代表材料呈韧性[34],BH/GH< 1.75代表材料呈脆性(金刚石B/G=0.8)。从表2中可以明显看出,若常压下ZrBeO3能稳定存在,其应为脆性材料,随着等静压力增大,材料逐渐向韧性转变。
2.3 声子能谱及热动力学稳定性
图3给出了晶胞结构优化后的ZrBeO3声子能谱,每单位晶胞有5个原子,因此有15支晶格振动格波模式。由于钙钛矿构型的体心高对称性,在G点(体心Be原子位置)15支格波呈共5组简并态,而在Q点(两Zr原子连线中点)格波谱线开裂间距最大[35],由Be和Zr原子质量差别较大所致,很明显声学支的晶格振动格波存在较多虚频(软化模式[36]),说明晶格整体在低温零压下处于热动力学不稳定状态,这可能是到目前为止实验未合成出该化合物的原因。晶胞在不同压力下的声子能谱如图4所示。可以看出,随着压力增大,声子虚频逐渐减小,等静压加到200 GPa时声子虚频消失。在273 K的温度下,对晶胞做了常压和200 GPa的动力学模拟(模拟动画见期刊官网的资源附件),结果显示200 GPa压力下晶格振动稳定,所以可认为ZrBeO3为高压生成稳定相。由于生成压力条件极高,因此实验室条件无法人工合成,但深层地壳或地核中可能存在该化合物。根据热力学稳定但动力学不活泼特性物质一般能形成力学强度高的材料[37],可以预测该化合物的力学强度很高。
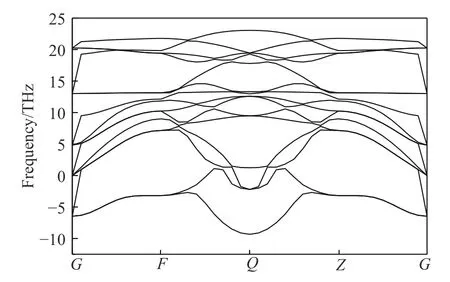
图3 零压下的ZrBeO3声子能谱Fig.3 ZrBeO3 phonon spectra at zero pressure
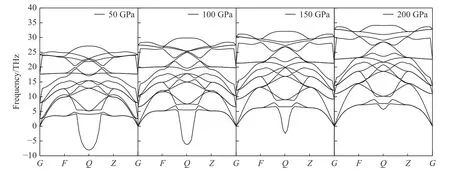
图4 不同压力下的ZrBeO3声子能谱Fig.4 Phonon spectra of ZrBeO3 at different pressures
表3对比了不同压力下ZrBeO3原子轨道及化学键布居值,可以从化学成键上说明在加压情况下ZrBeO3如何趋向于稳定。Be原子的s轨道布居值随等静压力增加而逐渐减小,p轨道布居值从1.11开始随压力增大而增加,说明钙钛矿结构ZrBeO3若要形成,Be原子不能处于基态,需要有s轨道的电子跃迁到p轨道,形成s-p杂化后与O原子成键,这可能是低压下ZrBeO3不能稳定的根本原因;同时随着压力增大,Be-O键布居值逐渐增加,键长逐渐减小,说明共价键属性增强,Be-O之间结合更紧密,从而使得八面体结构更稳定。而对于Zr原子,其s、p轨道布居值随压力增大而减小,d轨道布居值增加,Zr-O键布居值和键长均减小,说明压力增加过程中,Zr-O逐渐向离子键趋势转变,电荷更多集中到O原子附近以Be-O共价键形式存在,加强稳固Be、O形成的八面体结构,Zr原子更多地起到“填隙”作用,使得钙钛矿结构稳定。
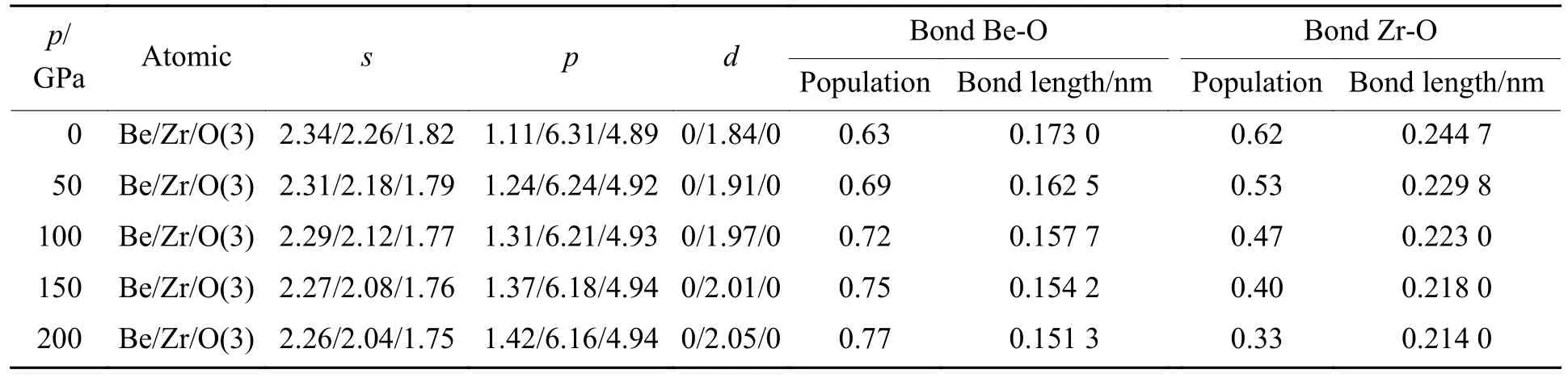
表3 不同压力下原子轨道和化学键的布居值分布Table 3 Atomic orbital and chemical bond population distribution at different pressures
为更清楚地显示压力对原子间相互作用的影响,给出不同压力下的电子总态密度及Be、Zr、O分波态密度图,如图5所示。在电子总态密度图中可以很容易看出,随着压力增大,电子总态密度向高能态方向增加,且每个能带宽度均逐渐增大,尤其在高能量区域(8~28 eV)的s和p电子最为明显,说明压力增大过程中,电子态密度有整体向高能级轨道跃迁的趋势,电子之间的相互作用逐渐增强,尤其是s-p电子之间。
从Be原子的电子分波态密度图中,能观察出其高能量区域中电子s-p杂化随着压力增大越来越明显。O原子分波态密度总体随压力变化不特别明显,高能区域变化趋势与总态密度相同,而在低能区域有个小波峰逐渐变大,表明该能量区域的电子密度有所增加(总体来说成分较小),可认为是Zr-O逐渐向离子键趋势转变促成。从过渡金属Zr原子分波态密度图中发现,随着压力增大,s轨道和d轨道出现分裂,这与价壳层中存在未配对电子有关。Be-O、Zr-O化学键的状态可以用“赝能隙”[38](Pseudogap,即在费米能级两侧分别有两个尖峰,而两个尖峰之间的态密度并不为零)判断,一般认为赝能隙宽度直接反映共价键强弱,越宽说明共价性越强。Be-O键的赝能隙宽度取决于O原子价带顶的p电子到Be原子导带底的p电子分布的距离,Zr-O键宽度取决于O原子价带顶的p电子到Zr原子导带底的d电子分布的距离。从图5(b)~图5(d)中数值标注可读出,Be-O键的共价性随压力增大而明显增强;Zr-O键共价性质却由于Zr的d轨道随着压力增大逐渐出现未配对电子情况而变得相对复杂,其赝能隙先增大后因轨道分裂出现子峰而减小,而轨道分裂意味着出现未配对电子,未配对电子的出现应是O原子吸收了Zr原子外围的价电子所致(即Zr-O键的离子性质随压力增大而逐渐增强)。
2.4 晶体电子能带结构及光学特性
计算了优化后晶胞的电子能带结构,能带结构图说明该化合物是具有间接能隙1.918 eV的半导体材料;优化后的晶胞的光学特性计算(主要关心可见光附近波段)结果表明,该材料在紫外段300 nm、可见光段500 nm、红外段700~900 nm有较强的吸收峰,可见光段600 nm附近处有最低的反射率。计算了该材料的介电常数随入射光能量的变化频率谱线,结果表明若能合成稳定化合物,其在半导体器件、光传感、光催化领域均具有较大潜力。考虑到材料的极不稳定性,相关计算结果的数据意义不大,暂作为支撑材料附上,见附录A。
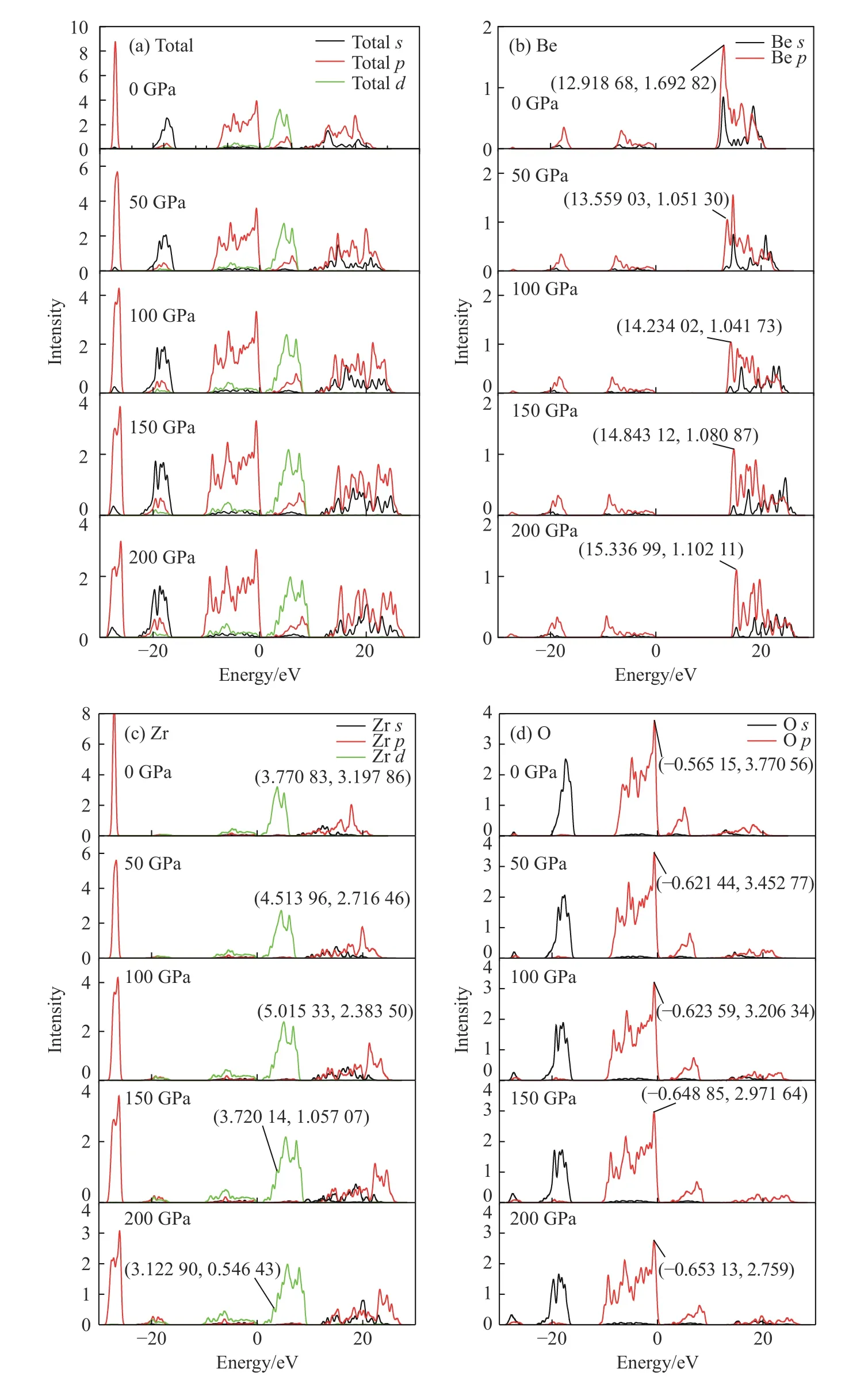
图5 不同压力下ZrBeO3电子总态密度及各原子分波态密度图Fig.5 Total density of state of ZrBeO3 electrons and partial density of states of each atom at different pressures
3 结 论
在满足钙钛矿几何结构稳定性、热力学机械稳定性的基础上,基于密度泛函理论(DFT)框架,使用广义梯度近似平面波方法(GGA)构建了钙钛矿构型ZrBeO3具体的晶体模型。结合能计算表明,该构型具有热力学稳定性;弹性常数、体积模量、剪切模量、杨氏模量的计算表明,该晶体具备较高的力学强度;硬度计算表明,该结构晶体硬度可与SiC相比拟;不同压力下的泊松比、BH/GH计算表明,材料在压力增加的情况下会由脆性向韧性转变。计算了ZrBeO3的声子能谱,结果表明ZrBeO3在低温零压下热动力学不稳定。为此对比分析了不同压力下的声子能谱、不同原子轨道和化学键布居值,晶胞的电子态密度及各个原子的电子分波态密度研究表明随着压力增加,Be原子sp杂化后形成的Be-O共价键成分增强、Zr-O键离子键成分增强,在200 GPa附近时晶格动力学趋于稳定。
附录A
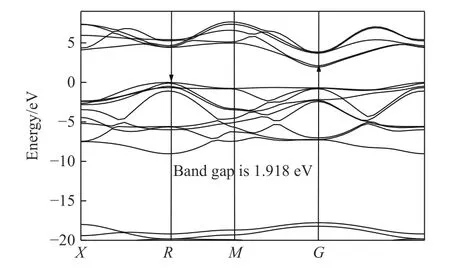
图A1 ZrBeO3的电子能带结构Fig.A1 Electronic band structure of ZrBeO3
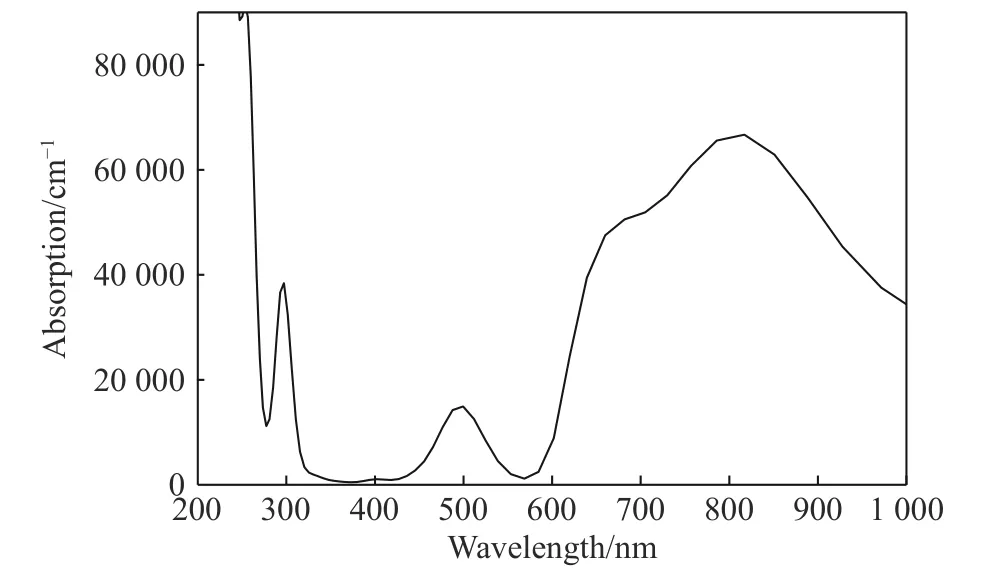
图A2 ZrBeO3的吸收谱Fig.A2 Optical absorption spectrum of ZrBeO3
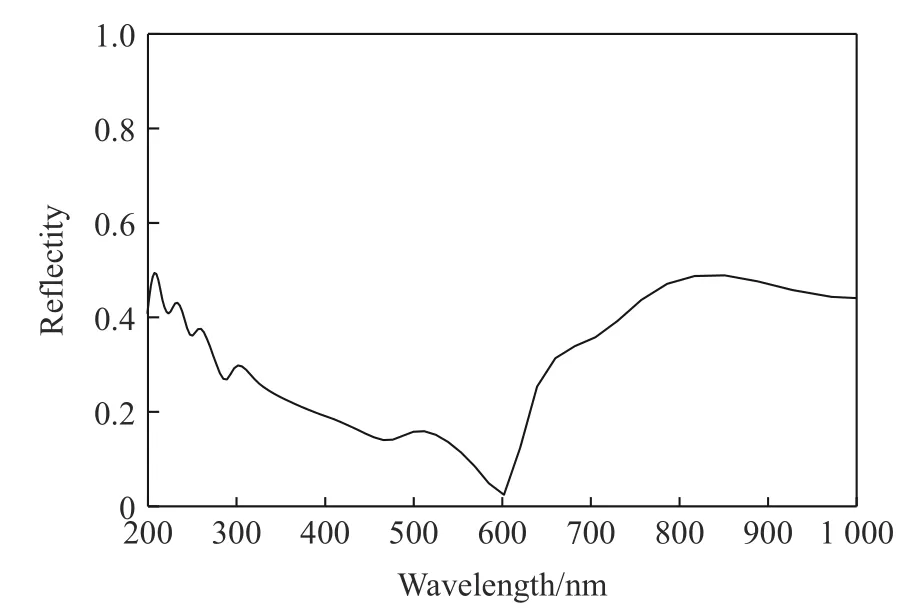
图A3 ZrBeO3的反射谱Fig.A3 Optical reflectivity spectrum of ZrBeO3
图A4 ZrBeO3介电函数谱线Fig.A4 Dielectric function spectrum of ZrBeO3
