金属氧化物改性的氧化锰八面体分子筛整体式催化剂催化分解臭氧性能*
2020-02-25陈崇来张文霞余会发贾爱平罗孟飞
陈崇来 张文霞 余会发 贾爱平 罗孟飞#
(1.金华职业技术学院制药与材料工程学院,浙江 金华 321017;2.浙江师范大学化学与生命科学学院,浙江 金华 321004)
臭氧具有强氧化性,能造成材料褪色、照片褪光、轮胎老化,导致植物坏死、生长受阻、作物减产,对人体组织的破坏性也很强[1-2]。目前,分解臭氧的方法有活性炭法、热分解法、药液吸收法和催化分解法[3-7]等,其中催化分解法因具有分解效率高,安全实用,又经济有效等优点,是分解臭氧较理想的方法。然而,催化剂种类繁多,制备方法也不尽相同,而进一步提高催化剂的臭氧分解性能是当前该领域中一项具有挑战性的研究课题。
根据活性组分的类型,臭氧分解催化剂可分贵金属(如Au、Pt、Pd等)催化剂[8-9]和非贵金属(如锰氧化物)催化剂[10]。前者由于价格昂贵限制了其大规模使用。锰氧化物不仅表现出优良的臭氧催化分解活性,还具备经济实用、效率高、使用方便等优点。其中氧化锰八面体分子筛(OMS-2)作为一种特殊的MnO2材料,因具有独特的[MnO6]八面体共边组成双链的隧道结构而引起了人们的关注[11]。为了进一步改善其结构特性及催化性能,通常在OMS-2中掺杂金属离子或者金属氧化物对其结构和催化性能进行改性。根据有关报道,可通过金属离子取代骨架或者隧道中的锰离子对OMS-2的催化性能进行调节。不同离子掺入到OMS-2隧道结构中与单纯的OMS-2催化剂相比较,不仅促进了氧缺位的形成,还提高了晶格氧(OⅠ)流动性,因而有利于臭氧的吸附和催化分解。用于臭氧分解的催化剂主要是通过在载体上浸渍活性组分(如锰氧化物),并且尽可能地分散活性组分,从而提供更多的反应活性中心。目前在文献中报道关于锰氧化物负载最多的载体为γ-Al2O3、活性炭,SiO2和沸石分子筛等作为载体也有所使用,但以堇青石蜂窝陶瓷作为载体负载锰氧化物较为少见,而锰氧化物本身具有多种混合价态、丰富的OⅠ以及优良的催化分解臭氧性能,所以将锰氧化物负载在堇青石蜂窝陶瓷制成整体式催化剂开展研究具有重要意义。
本研究通过制备一系列不同金属氧化物改性的OMS-2整体式催化剂,并对催化剂进行臭氧分解性能评价和结构表征,建立催化剂的构效关系,为制备性能优良、经济实用的整体式臭氧分解催化剂提供一定的理论基础。
1 实验部分
1.1 催化剂的制备
1.1.1 OMS-2粉末的制备
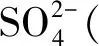
1.1.2 不同金属氧化物改性的OMS-2整体式催化剂的制备
按照金属氧化物Fe2O3、Co3O4、NiO、CuO和CeO2与OMS-2质量比为1∶4的掺杂比例,分别称取不同金属氧化物的前驱体盐Fe(NO3)3·9H2O、Co(NO3)2·6H2O、Ni(NO3)2·6H2O、Cu(NO3)2·3H2O、Ce(NO3)3·6H2O(均为分析纯)和OMS-2粉末,加蒸馏水混合,在常温常压下分别与黏结材料铝胶相互混合,并用球磨机研磨2 h,然后将研磨好的浆液涂覆到堇青石蜂窝陶瓷载体上,400 ℃焙烧3 h,最终制得整体式催化剂,并命名为M-OMS-2催化剂(M为掺杂的金属元素,即Fe、Co、Ni、Cu、Ce)。涂覆后剩余的浆液在120 ℃烘干过夜,400 ℃焙烧3 h后得到的M-OMS-2粉末(不含载体)用于催化剂表征。
1.2 催化剂的表征
1.2.1 X射线粉末衍射(XRD)分析
采用德国Bruker D8ADVANCE型XRD仪对催化剂的晶相结构进行测定,其实验条件为:以Cu Kα辐射为激光光源(波长为0.154 18 nm),仪器管电压40 kV,管电流40 mA,扫描速度0.067°/s,扫描范围10°~90°。
1.2.2 拉曼光谱分析
利用英国Renishaw inVia型拉曼光谱仪对催化剂进行拉曼光谱分析。实验条件为:激光波长532 nm,扫描范围200~1 200 cm-1,保留时间60 s,扫描4次,分辨率1 cm-1。
1.2.3 X射线光电子能谱(XPS)分析
催化剂中各个元素的化学态及相对含量测试是在美国Thermo Fisher ESCALAB250Xi型XPS仪上进行的。使用单色器Al Kα射线(1 486.6 eV)采集数据,结合能通过样品表面污染物C 1s(284.8 eV)校正。
1.2.4 BET比表面积和孔结构的测定
采用JW-BK122F型比表面及孔径分析仪测定催化剂的比表面积和孔结构。在进行氮气吸附之前需要将干燥的催化剂样品在220 ℃下脱气至0.01 Pa。通过BET方法计算得到催化剂的比表面积,再通过BJH方法计算吸附/脱附等温线,得出孔容、孔径及其分布等孔结构信息。
1.2.5 扫描电子显微镜(SEM)观察
利用日本Hitachi S-4800型SEM观察催化剂的形貌。
1.2.6 程序升温氢气还原(H2-TPR)分析
在自制的微型反应装置上进行了H2-TPR实验,实验流程如下:将50 mg催化剂粉末置于石英管(管径6 mm)中间处并用石英棉固定,将K型热电偶装在催化剂处的石英管外壁;通入经脱氧和分子筛脱水净化处理的氢气(体积分数5%)-氮气(体积分数95%)混合气,流速为30 mL/min;热导基线稳定后,从室温以10 ℃/min的升温速率对样品进行程序升温至800 ℃并用软件记录,其中氢气的变化通过热导检测器(TCD)检测。以纯0.05 g CuO作为标准样,得到CuO和峰面积的比值作为校正因子,从而计算催化剂的耗氢量。
1.3 催化剂活性评价
催化剂活性评价在内径为16 mm的固定床反应器中进行。评价条件为:常温常压下,臭氧入口质量浓度70.62 mg/m3,空速21 600 h-1,气体流量0.5 m3/h,催化剂用量14 g。采用自制的紫外线灯臭氧发生器产生臭氧,反应过程中臭氧浓度实时测量,分别计算不同催化剂的臭氧转化率和反应速率。
2 结果与讨论
2.1 比表面积和孔结构
从表1中可以看出,不同金属氧化物改性对OMS-2催化剂的比表面积、孔容及孔径均产生一定程度的影响。其中与未改性的OMS-2催化剂相比,Fe-OMS-2催化剂比表面积增加最明显,而Co-OMS-2催化剂比表面积下降程度最大。
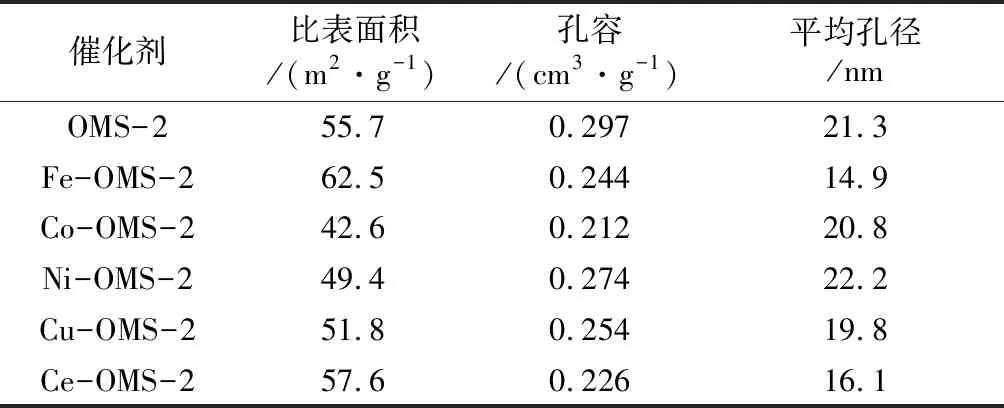
表1 M-OMS-2催化剂的比表面积和孔结构
2.2 活性评价
从图1可以看出,不同金属氧化物进行改性对催化剂性能产生了影响。Cu-OMS-2催化剂在6 h内对臭氧的转化率能够保持在100.0%,其活性略高于OMS-2催化剂。然而其他金属氧化物改性则对OMS-2催化剂催化分解臭氧的活性起抑制作用。其中Co3O4改性对OMS-2催化剂臭氧分解活性的抑制作用最明显,经过6 h,臭氧转化率从最初的100.0%降至85.0%。而Ni-OMS-2和Ce-OMS-2催化剂经过2 h,其反应活性略微有所下降。反应6 h时,臭氧转化率及反应速率均为Cu-OMS-2催化剂>OMS-2催化剂>Ni-OMS-2催化剂>Ce-OMS-2催化剂>Fe-OMS-2催化剂>Co-OMS-2催化剂(见表2),表明CuO改性的OMS-2催化剂比未改性的OMS-2催化剂表现出更好的臭氧分解活性,而其他金属氧化物改性的OMS-2催化剂的活性均有一定程度降低。结合表1、表2来看,不同催化剂作用下的反应速率与催化剂比表面积不存在对应关系,说明有其他因素影响了催化剂的反应活性。
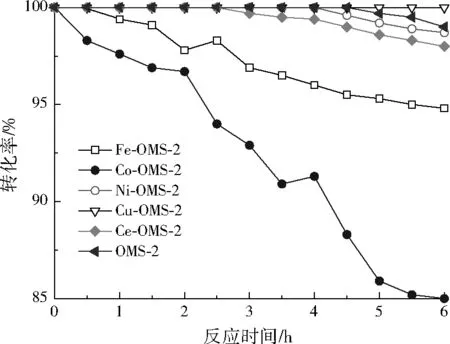
图1 M-OMS-2催化剂的臭氧分解活性随反应时间的变化Fig.1 The catalytic activity of M-OMS-2 catalysts for ozone decomposition varied with time
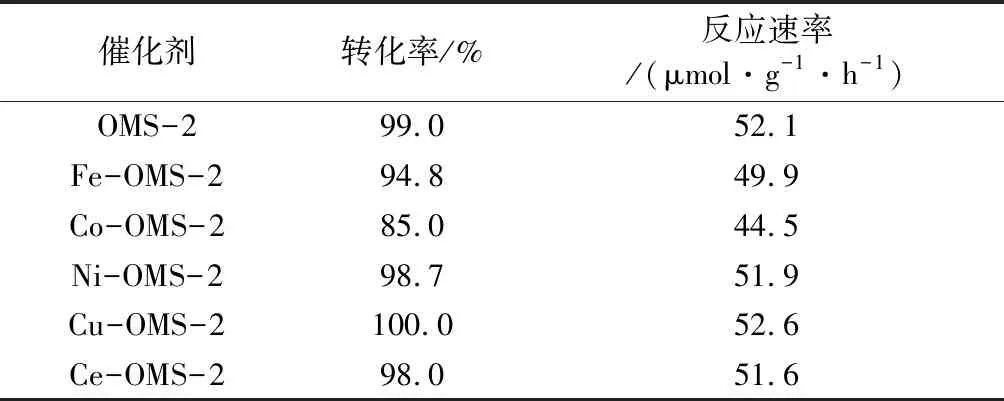
表2 M-OMS-2催化剂的臭氧转化率和反应速率1)
注:1)臭氧转化率和反应速率均针对反应时间6 h进行计算。
2.3 催化剂的物相结构
从图2可以看出,M-OMS-2催化剂都出现了归属于OMS-2(JCPDS29-1020)的锰钾矿结构特征衍射峰[12],表明改性前后OMS-2基本结构保持不变。同时可以看出M-OMS-2催化剂的OMS-2峰宽略有不同,OMS-2颗粒尺寸略有差别。此外,M-OMS-2催化剂还有归属于所掺杂的金属氧化物的衍射峰。
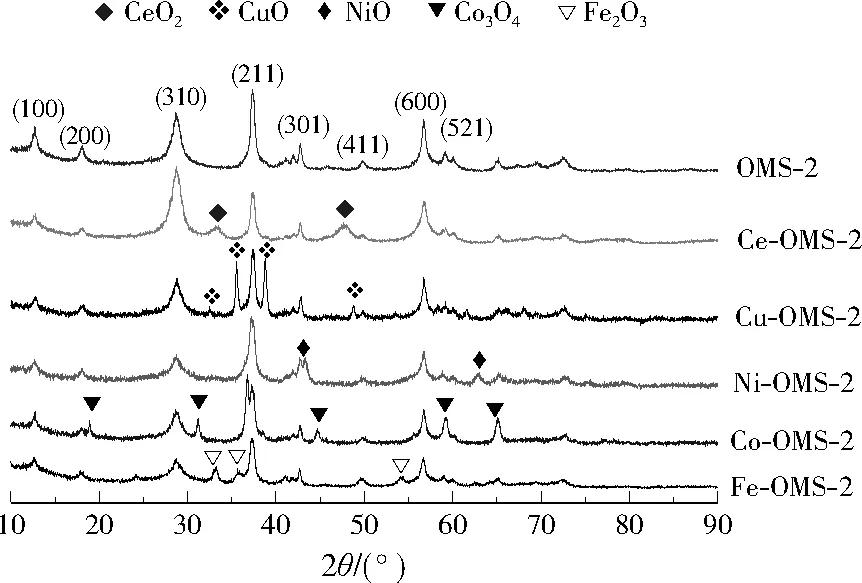
图2 M-OMS-2催化剂的XRD图谱Fig.2 XRD patterns of M-OMS-2 catalysts
通常在掺杂金属氧化物的OMS-2催化剂中,金属元素可能有两种不同存在形式[13]:一是以金属离子的状态存在于OMS-2晶胞中;另一种是以自由态氧化物的形式存在,没有进入OMS-2晶胞。图2表明,M-OMS-2催化剂中的金属元素并未完全掺入OMS-2晶胞中,部分以自由态氧化物存在。
2.4 形貌特征
从图3可以看出,M-OMS-2催化剂都呈颗粒状并夹杂着棒状物。与未改性的OMS-2催化剂相比,金属氧化物改性对OMS-2催化剂形貌的影响不明显。

图3 M-OMS-2催化剂的SEM图(×100 000)Fig.3 SEM images of M-OMS-2 catalysts (×100 000)
2.5 拉曼光谱
从图4可以看出,所有M-OMS-2催化剂都在638 cm-1附近出现了归属于[MnO6]八面体双链上Mn—O晶格的垂直伸缩振动造成的拉曼振动峰[14],这说明了金属氧化物改性未对OMS-2催化剂的结构产生明显影响,与XRD结果吻合。但是,M-OMS-2催化剂的振动峰强度有所变化,说明金属氧化物改性引起了Mn—O晶格化学环境的变化。除此之外,Ni-OMS-2在530 cm-1处出现了归属于NiO的振动峰,说明NiO在OMS-2催化剂表面有一定程度的富集。而在其他M-OMS-2催化剂中,金属氧化物的拉曼振动峰不明显,说明这些金属氧化物大部分存在于M-OMS-2催化剂中。
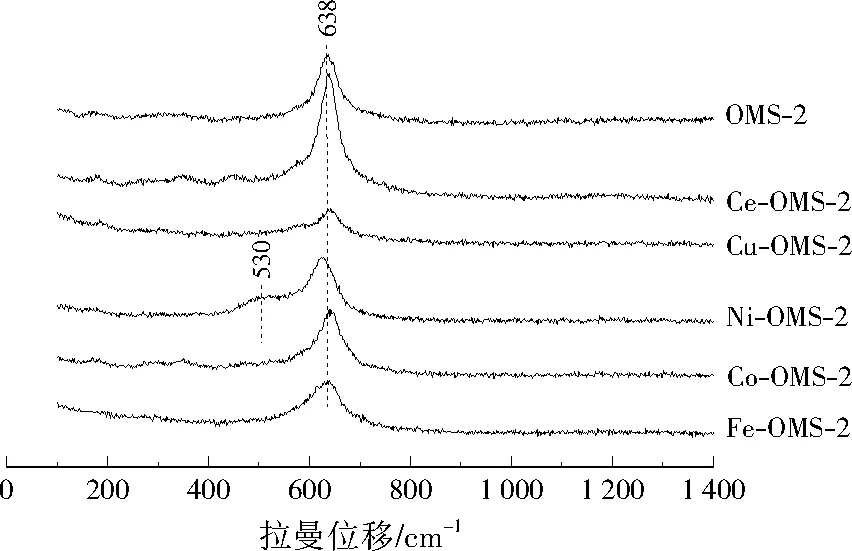
图4 M-OMS-2催化剂的拉曼光谱Fig.4 Raman spectra of M-OMS-2 catalysts
2.6 XPS表征
图5展示了M-OMS-2催化剂的Mn 2p和O 1s图谱。一般认为,641.4、641.9、642.4 eV左右的结合能分别归属于Mn2+、Mn3+和Mn4+。根据M-OMS-2催化剂中Mn 2p3/2结合能位置判断,大部分M-OMS-2催化剂中Mn 2p3/2的结合能基本都在642.4 eV左右,说明Mn主要以Mn4+的形式存在,而Cu-OMS-2中可能含有较多含量的Mn3+。利用Mn 3s的分裂能级差可以较准确地判定Mn价态,计算出Mn的平均氧化价态(AOS)[15],结果列于表3。CuO改性对OMS-2催化剂中Mn的价态影响最明显,其中Mn基本以Mn3+为主,而较高的Mn3+含量能促进臭氧分解活性[16]。这可能归因于Cu与Mn之间显著的电子交换作用。虽然其他金属氧化物改性也造成了OMS-2中Mn价态下降,但是催化剂活性却受到抑制,说明还有其他因素的影响。
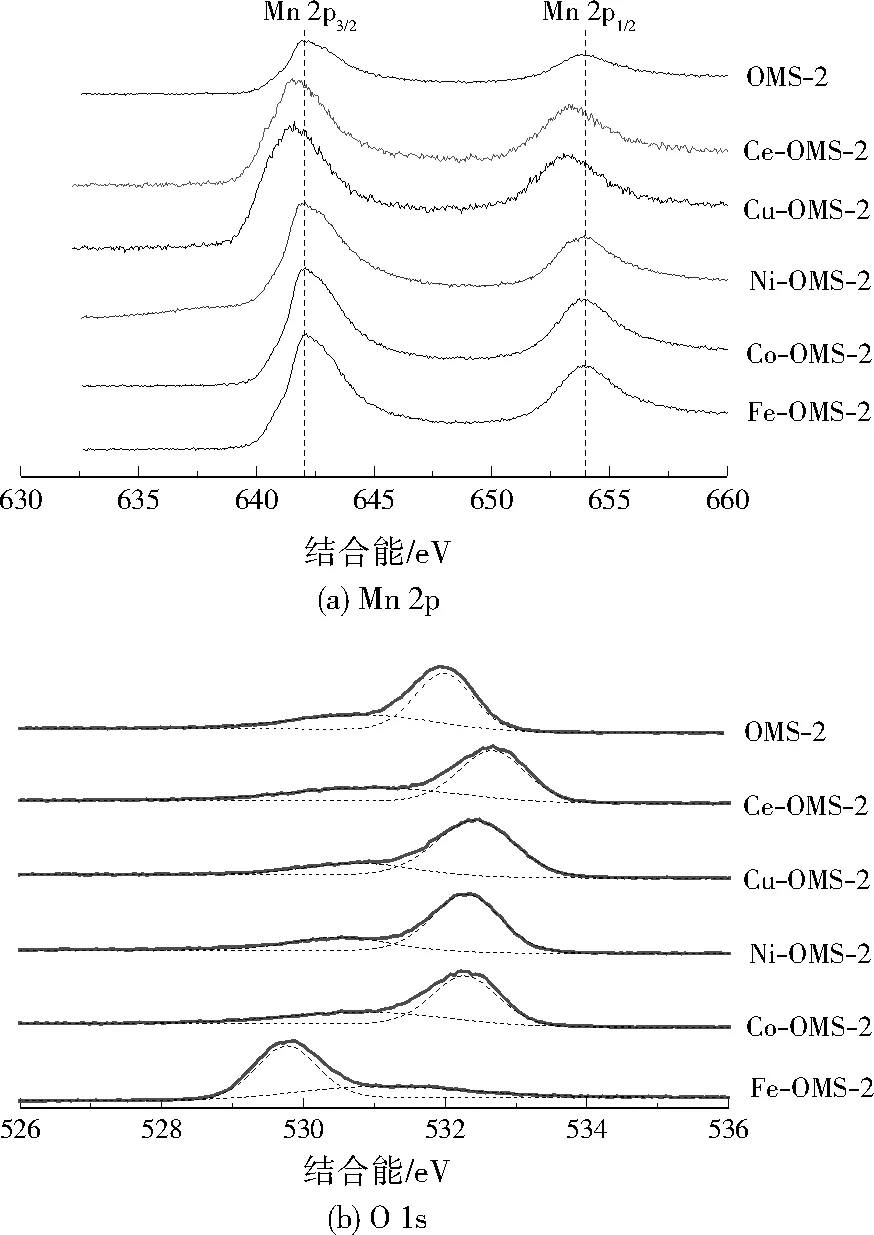
注:图5(b)中的虚线为分峰拟合线。图5 M-OMS-2催化剂的XPS图谱Fig.5 XPS spectra of M-OMS-2 catalysts
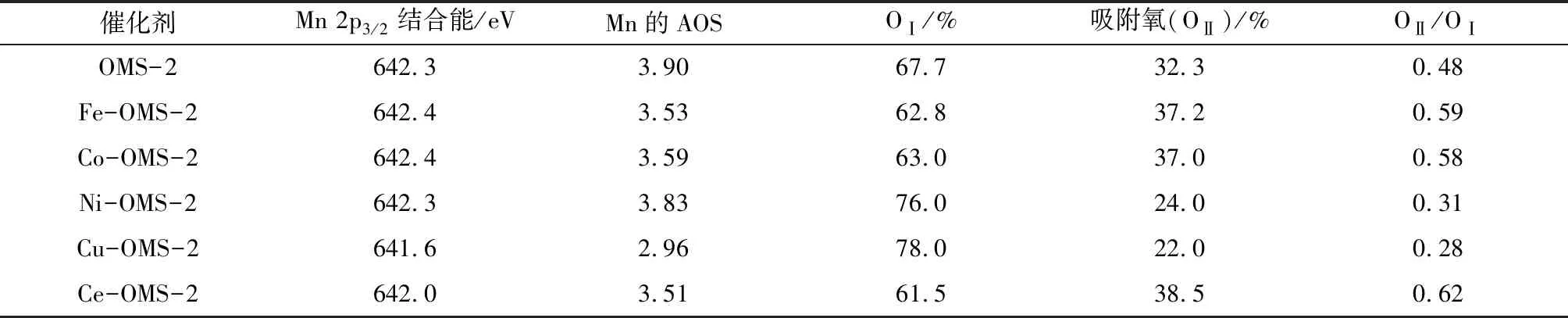
表3 M-OMS-2催化剂的XPS图谱分析结果1)
注:1)OⅠ和OⅡ占比均以摩尔分数计;OⅡ/OⅠ为摩尔比。
对O 1s图谱进行分峰,529.5~529.7 eV归属于OⅠ,531.0~531.2 eV归属于OⅡ。根据表3可以看出,Cu-OMS-2的氧缺位含量(以OⅡ/OⅠ表征)最少,而Ce-OMS-2的氧缺位含量最多,表明氧缺位含量并不是决定催化剂活性的主要因素。催化剂中的氧缺位和催化剂的失活机理密切相关。根据图1结果,除了Co-OMS-2、Fe-OMS-2催化剂较早失活以外,OMS-2、Ce-OMS-2和Ni-OMS-2催化剂在反应3~5 h活性也开始先后下降。臭氧在催化剂表面的分解机理一般认为是臭氧与催化剂表面的氧缺位结合以后,生成过氧化物,接着过氧化物分解,然后释放氧气,氧缺位复原,继续参与下一个反应。如果催化剂表面的过氧化物不能及时分解,容易转变为晶格氧,会造成Mn的氧化价态升高,即Mn3+逐渐转化为Mn4+。Cu-OMS-2相比其他M-OMS-2催化剂Mn3+含量最多,从而最不容易失活。
2.7 还原性能
从图6可以看出,大多数M-OMS-2催化剂的还原峰由3个小峰叠加而成,分别归属为MnO2到Mn2O3、Mn2O3到Mn3O4及Mn3O4到MnO的还原[17-18]。还原峰的特征还与M-OMS-2催化剂的结构有关。一般认为臭氧在锰氧化物上的催化分解主要包括两个步骤[19]:第1步是臭氧在催化剂表面的吸附;第2步是吸附中间体的脱附。在这过程中臭氧发生还原反应,如果催化剂越容易被氧化,其对应的臭氧分解速率也就越快。从表4可以看到,M-OMS-2催化剂具有不同的还原温度和耗氢量。其中Cu-OMS-2催化剂相比未改性的OMS-2催化剂,还原温度降低,耗氢量升高,表明Cu-OMS-2催化剂具有更好的还原能力,因此相应的臭氧转化率更高,这可能也是Cu-OMS-2催化剂具有最好反应活性的根本原因。虽然Co-OMS-2和Ni-OMS-2催化剂的耗氢量升高,但是其还原温度也明显升高;而Fe-OMS-2和Ce-OMS-2催化剂还原温度升高且耗氢量降低,导致其反应活性受到了抑制。H2-TPR分析结果验证了催化剂的活性测试结果,表明催化剂的还原能力是决定其活性的关键因素。
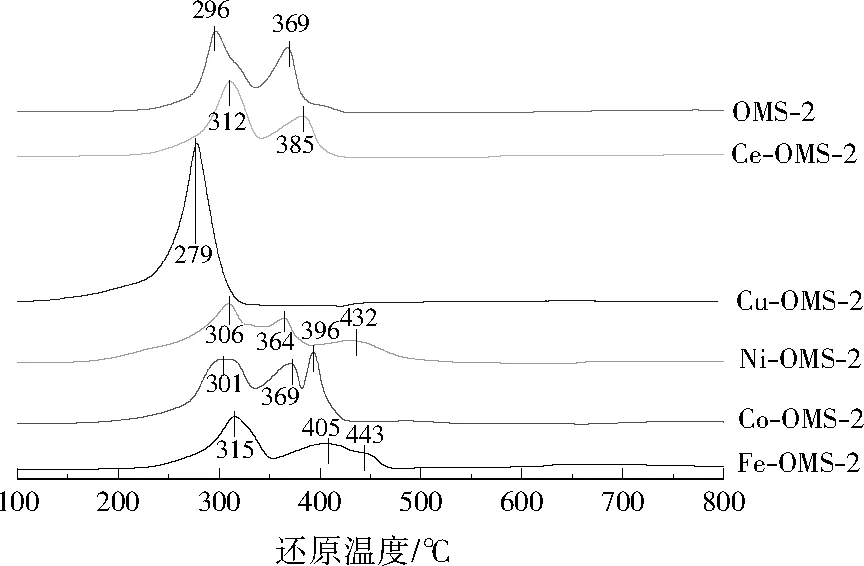
图6 M-OMS-2催化剂的H2-TPR图Fig.6 H2-TPR profiles of M-OMS-2 catalysts
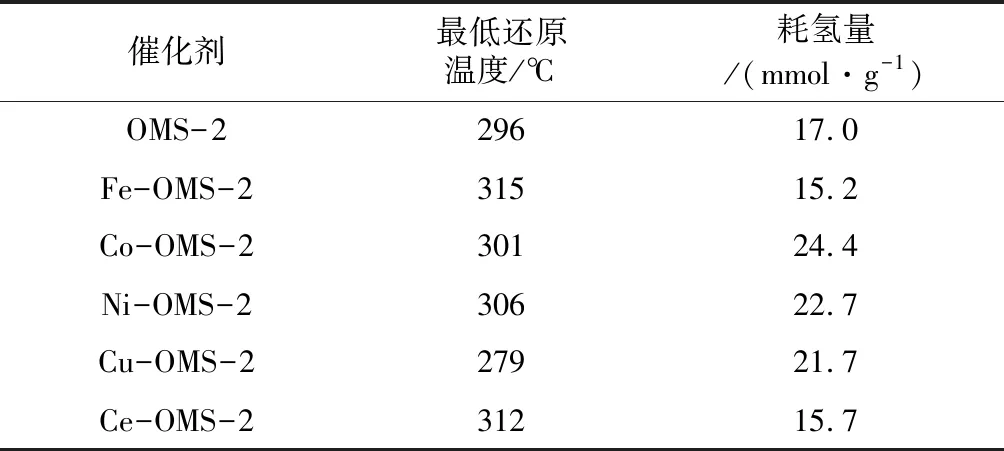
表4 M-OMS-2催化剂的最低还原温度和耗氢量
3 结 论
(1) Cu-OMS-2催化剂表现出比未改性的OMS-2催化剂更高的反应速率,而其他M-OMS-2催化剂反应速率有一定程度降低。
(2) 不同金属氧化物改性未对OMS-2催化剂的物相结构和形貌产生明显影响。
(3) 金属氧化物改性改变了OMS-2表面的Mn价态和还原性能。其中CuO改性提高了OMS-2催化剂表面的Mn3+浓度和还原性能,从而有利于提高其臭氧分解活性。
