CoMo/MgO催化剂的制备及其对二苯并噻吩的加氢脱硫性能
2020-02-21郑世富黄佩佩
郑世富,黄佩佩
(江西赣锋锂业股份有限公司,江西 新余 338000)
随着环境法规和人们环保意识的加强,超低硫含量燃油成为人们关注的热点[1]。加氢脱硫技术(HDS)是实现石油馏分脱硫最行之有效的方法。传统的HDS催化剂为氧化铝(γ-Al2O3)担载的CoMo硫化物催化剂[2-3]。然而,γ-Al2O3负载的HDS催化剂对大分子含硫化合物(DBT、4,6-DMDBT等)的脱硫活性较低,不能实现超低硫含量燃油的生产。因此,高效HDS催化剂是实现燃油深度脱硫最有效的方法。
研究表明,HDS活性很大程度上取决于载体的特性,比如:载体表面的化学特性。为了提高HDS的性能,研究者们对γ-Al2O3的表面性能进行了改进。例如:将磷(P)、氟(F)作为添加剂引入γ-Al2O3载体中,改变催化剂的酸度,提高硫化物活性相的比例,从而提高催化剂的HDS性能。Han等[4]研究发现,B酸可以使活性相之间形成电子缺陷,从而增加催化剂的加氢和氢解活性。另一方面,通过改变催化剂载体制备新型HDS催化剂,比如:ZSM-5[5]、TiO2[6]、Al-MCM-41[7]。然而,上述载体大部分为酸性载体,酸性载体在反应的过程中,容易形成积碳,降低催化剂的活性[8]。
近年来,采用溶胶-凝胶法制备含氧化镁(MgO)的二元氧化物载体应用在HDS反应上,表现出了很高的HDS活性,这主要归功于活性相分散度的提高。Mogica-Betancourt等[9]以γ-Al2O3-MgO为载体,制备NiW硫化物催化剂,结果表明,向γ-Al2O3载体中加入MgO,可以直接增强W与载体间的相互作用,促进W物种的分散,使W易于硫化,从而提高催化剂的活性。此外,在二元氧化物中存在的MgO可以有效抑制非活性相CoAl2O4和Co3O4的形成[10]。Chary等[11]通过实验发现,二硫化钼(MoS2)在MgO载体中的分散性较高,所以其HDS活性远高于传统载体γ-Al2O3担载的MoS2催化剂。与此同时,MgO载体独特的物化性质,使得其所担载的金属硫化物催化剂具有良好的耐氮性和稳定性,因此,MgO载体有望成为新型的HDS载体。
笔者采用MgO为载体,制备负载型CoMo催化剂(CoMo/MgO),考察其对DBT的催化性能。作为比较,进一步以γ-Al2O3为载体,制备CoMo/γ-Al2O3催化剂,并研究不同催化剂上Mo物种的硫化程度和金属活性相的分散状态。
1 实验部分
1.1 试剂和原料
六水合硝酸钴(Co(NO3)2·6H2O)和四水合钼酸铵((NH4)6Mo7O24·4H2O),分析纯,国药集团化学试剂有限公司产品;二苯并噻吩(DBT,质量分数98%),纽安节化工科技有限公司产品;十氢萘(质量分数98%),成都联合化工有限公司产品;氢氧化镁(Mg(OH)2,质量分数99%),河北镁盛化工科技有限公司;氧化铝(γ-Al2O3,工业级),上海恒业分子筛股份有限公司产品。
1.2 氧化镁载体的制备
室温下,依次向Parr釜中加入3 g氢氧化镁和150 mL蒸馏水,将Parr釜密封。然后充入CO2气体置换Parr釜内空气,反复操作3次以上,以保证Parr釜内空气全部被排尽后,增压至5 MPa,并以200 r/min的转速搅拌3 h。反应结束后,排尽Parr釜内的气体,得到澄清透明的溶液。然后,将澄清溶液倒入烧杯中,在85 ℃水浴下连续搅拌20 min以上,析出晶体后经抽滤、洗涤、干燥后得到氧化镁前驱体,并在400 ℃下焙烧3 h(升温速率为3 ℃/min),得到MgO载体。
1.3 催化剂的制备
CoMo/MgO催化剂的制备采用等体积浸渍法:取1 g MgO载体,称取一定量的(NH4)6Mo7O24·4H2O和Co(NO3)2·6H2O,并溶于一定量的蒸馏水中,然后将所得溶液等体积浸渍到MgO载体上。样品在室温下放置12 h,100 ℃下干燥12 h,然后将干燥后的样品在10 MPa下压片成型、压碎过筛,得到40~60目的颗粒,所得催化剂前躯体于400 ℃煅烧2 h。
作为比较,以传统γ-Al2O3为载体,采用相同的方法负载CoMo催化剂,标记为CoMo/γ-Al2O3。
上述催化剂均在H2-H2S(20%体积分数H2S)混合气体中400 ℃硫化3 h,升温速率为2 ℃/min,气体流量为30 mL/min。其中,Co和Mo的负载质量分数均分别为3.3%和10.7%。
1.4 催化剂的表征
样品的X射线多晶粉末衍射(XRD)分析在RIGAKU Smart Lab衍射仪上完成,Cu靶Kα辐射,管电压40 kV,管电流100 mA,2θ扫描范围5°~80°。
氮气的吸脱附曲线在Micromeritics ASAP 2020M设备上获得。样品的表面积和孔径分布分别采用Brunner-Emmet-Teller(BET)方法和Barrett-Joyner-Halenda(BJH)方法计算。
紫外可见光谱漫反射(UV-vis)分析在UV-3600设备上完成。其中样品对光的反射值用R∞表示,所得数据通过Kubelka-Munk(F(R∞))函数[9]处理。
X射线光电子能谱分析(XPS)在ESCALAB MK Ⅱ设备上完成。
样品的透射电镜(TEM)照片在JEM-2100型设备上获得。通过大量的TEM照片,对MoS2活性相的堆垛层数和长度进行统计分析。假定MoS2活性相是规则的六边形,从而估算出MoS2的分散度(DMo)[12]。DMo定义为位于MoS2边缘表层Mo原子数所占总的Mo原子数的分数,可以采用公式(1)和(2)进行计算[13]
(1)
L=3.2(2ni-1)Å
(2)
式中,ni为MoS2相中边缘Mo原子的数量;t是统计TEM图片中MoS2条纹数目;L是MoS2的颗粒长度。
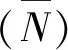
(3)
(4)
式中:Ni是所统计的第i条MoS2颗粒堆垛层数;yi是所统计的第i条MoS2颗粒堆积层数为Ni的数目;Li是所统计的第i条MoS2颗粒长度;xi是所统计的第i条MoS2颗粒长度为Li的数目。
1.5 催化剂活性评价
催化剂的活性测试在固定床反应装置上进行,取硫化后的催化剂0.2 g,采用石英砂作为稀释剂,DBT为模型化合物,十氢萘为溶剂,其中DBT的质量分数为0.57%。反应条件为:反应温度260 ℃、氢分压为3.0 MPa、氢气流量为50 mL/min、质量空速(WHSV)为19 h-1。反应稳定5 h后,每隔1 h采集一次,使用配备FID检测器的Agilent 7890B GC分析。
在消除质量传递和热量传递条件下,比较催化剂的本征加氢脱硫活性。实验采用0.05 g硫化的催化剂,与石英砂充分混合后装入反应器。反应条件为:温度为260 ℃、氢分压为3.0 MPa、氢气流量为50 mL/min、DBT的质量分数为0.57%、WHSV范围为50~160 h-1。
DBT 加氢脱硫的反应速率常数(kHDS)和转化频率(TOF)由公式(5)和(6)进行计算[15]:
(5)
(6)
式中,x为DBT的转化率,%;F为DBT的摩尔流量,mol/s;NMo为催化剂中Mo原子的物质的量,mol;m为催化剂的质量,g。
2 结果与讨论
2.1 催化剂的物性表征
2.1.1 XRD表征
图1(a)为CoMo/MgO、CoMo/γ-Al2O3催化剂煅烧后的XRD图谱。


图1 CoMo/MgO、CoMo/γ-Al2O3催化剂的XRD图谱
从图1(a)可知,在CoMo/MgO催化剂上,并未发现Co和Mo物种相关的特征衍射峰,这就说明金属很好的分散在MgO载体的表面;在CoMo/γ-Al2O3催化剂上,出现了MoO3(2θ=13.0°、23.3°、27.1°、33.6°)[16]和CoMoO4(2θ=14.1°、25.4°、28.3°、32.0°、36.6°、43.4°)[17]的特征衍射峰。催化剂经硫化后的XRD图谱见图1(b),从图1(b)可知,在CoMo/MgO催化剂上,并未发现Co和Mo物种相关的特征衍射峰;在CoMo/γ-Al2O3催化剂上,出现了MoS2(2θ=14.1°、33.6°、39.5°)[11]和Co3S4(2θ=26.8°、31.6°、38.2°、50.4°、55.2°)[5]的特征衍射峰。综上可知,金属活性相在CoMo/MgO催化剂上的分散性比CoMo/γ-Al2O3催化剂的好。
2.1.2 氮气吸附-脱附表征
图2为MgO载体、γ-Al2O3载体N2-吸脱附曲线和孔径分布。由图2(a)可知,MgO载体、γ-Al2O3载体的N2-吸脱附曲线均存在明显的滞后环,说明MgO载体和γ-Al2O3载体均具有介孔结构。但是,与MgO载体不同,γ-Al2O3载体的滞后环明显的向着高比压区偏移。因此,两个载体呈现了不同的孔径分布,MgO载体的孔径大小主要集中在3.5 nm,γ-Al2O3载体的孔径大小主要集中在7.6 nm。两种载体均负载上CoMo金属,经煅烧、硫化后得到的N2-吸脱附曲线、孔径分布见图2(b)。从图2(b)可知,MgO载体负载CoMo金属并经煅烧、硫化处理后,CoMo/MgO催化剂的滞后环明显的向着高比压区偏移,因此与MgO载体不同,CoMo/MgO催化剂的孔径大小主要集中在12.4 nm。然而,γ-Al2O3载体负载CoMo金属并经煅烧、硫化处理后,CoMo/γ-Al2O3催化剂的吸脱附曲线并没有引起明显的变化。
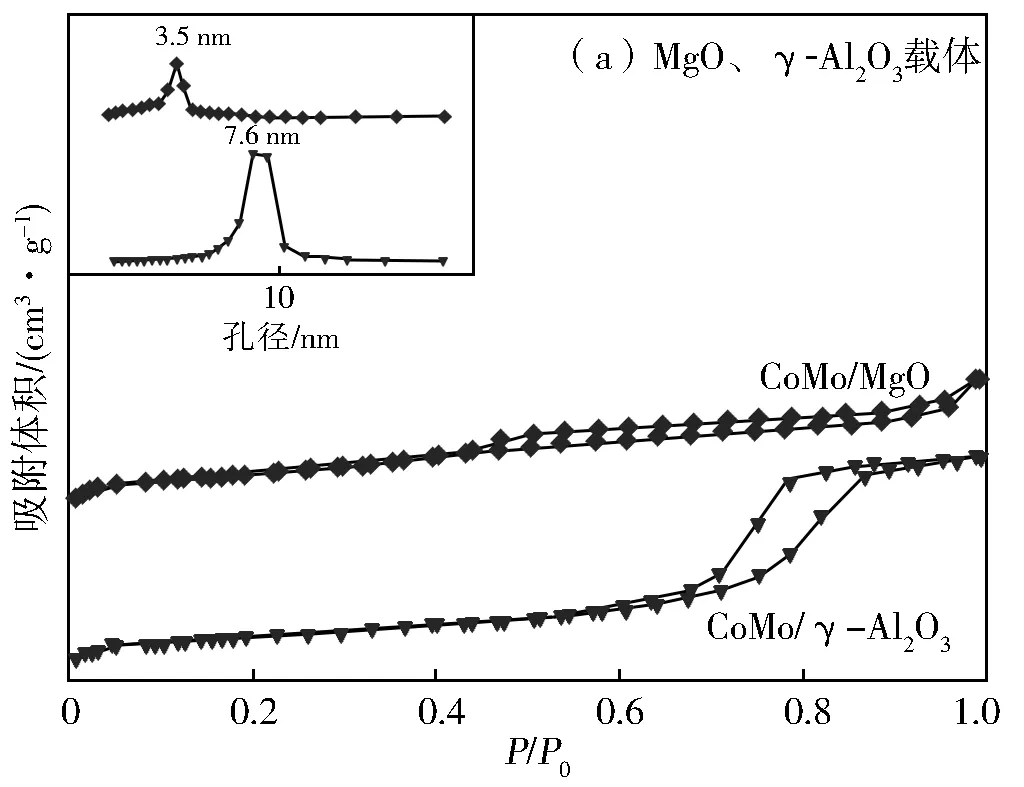
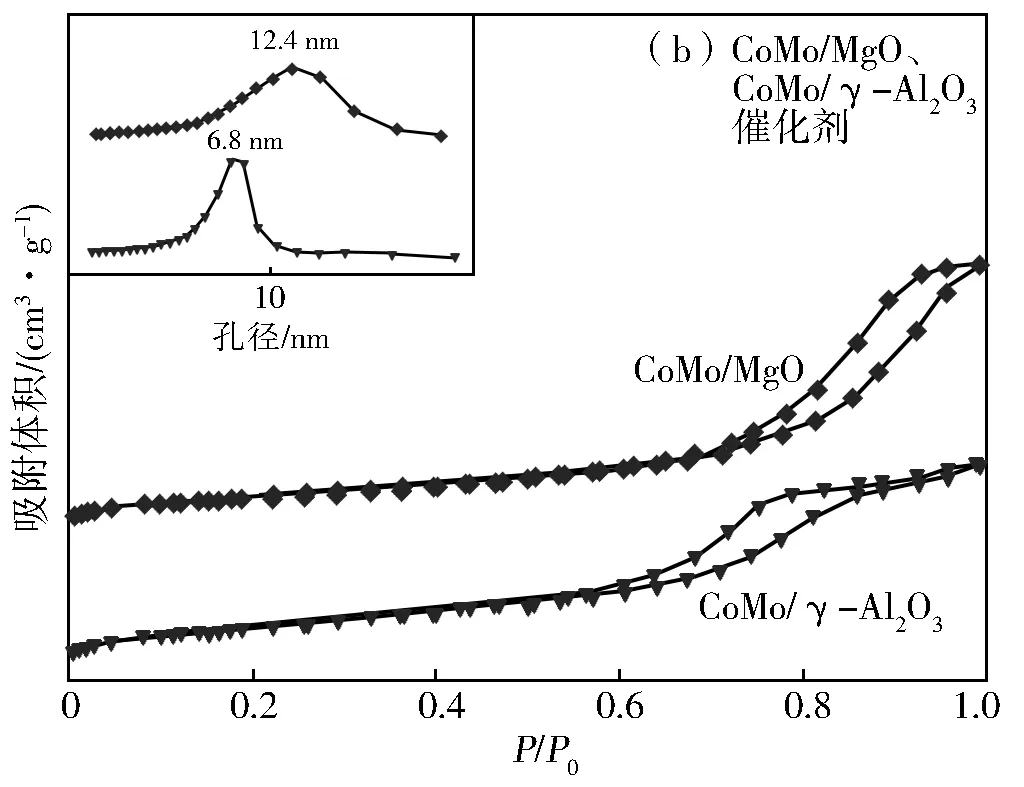
图2 N2吸脱附曲线和孔径分布
表1列出了MgO、CoMo/MgO、γ-Al2O3和CoMo/γ-Al2O3催化剂的比表面积、介孔体积和平均孔径大小。由表1可知,MgO载体、γ-Al2O3载体负载CoMo金属并经过煅烧、硫化处理后,催化剂的比表面积均有所降低,表明CoMo催化剂沉积到载体的表面或是孔结构中。其中,CoMo/MgO催化剂的介孔体积有所增加,这可能与催化剂制备过程中MgO与水发生反应有关。介孔体积越大,越有利于物质的传递。

表1 MgO、γ-Al2O3、CoMo/MgO和CoMo/γ-Al2O3的织构参数
2.1.3 UV-vis表征
采用UV-vis分析煅烧后催化剂Mo物种的配位状态,结果见图3。

图3 CoMo/MgO、CoMo/γ-Al2O3催化剂煅烧后的UV-vis谱
一般认为,200~400 nm的吸附带与O2-→Mo6+电荷转移、氧化钼物种的配位及聚集状态有关,230~260 nm的吸附带主要为四配位的Mo物种,270~330 nm的吸附带主要为六配位的Mo物种[14]。在CoMo/γ-Al2O3催化剂上吸收峰出现在220 nm附近,而在CoMo/MgO催化剂上,除了220 nm附近的吸收峰之外,280 nm附近也明显的观察到1个吸收峰。除此之外,CoMo/γ-Al2O3催化剂还在585 nm附近出现了1个吸收峰,这是因为生成了CoAl2O4物种[16]。因此,与CoMo/γ-Al2O3催化剂相比,在CoMo/MgO催化剂上形成了更多的六配位的Mo物种。众所周知,通过UV-vis数据,可以得到hv与(F(R∞)hv)2的线性相关曲线,从而得到禁带宽度(Eg),进一步计算得到钼物种的平均粒径,其中,钼物种的平均粒径与Eg负相关。由图4计算出CoMo/γ-Al2O3催化剂的Eg为3.1 eV,CoMo/MgO-400催化剂的Eg为3.4 eV。因此,与CoMo/γ-Al2O3催化剂相比,CoMo/MgO催化剂上钼物种的平均粒径较小。
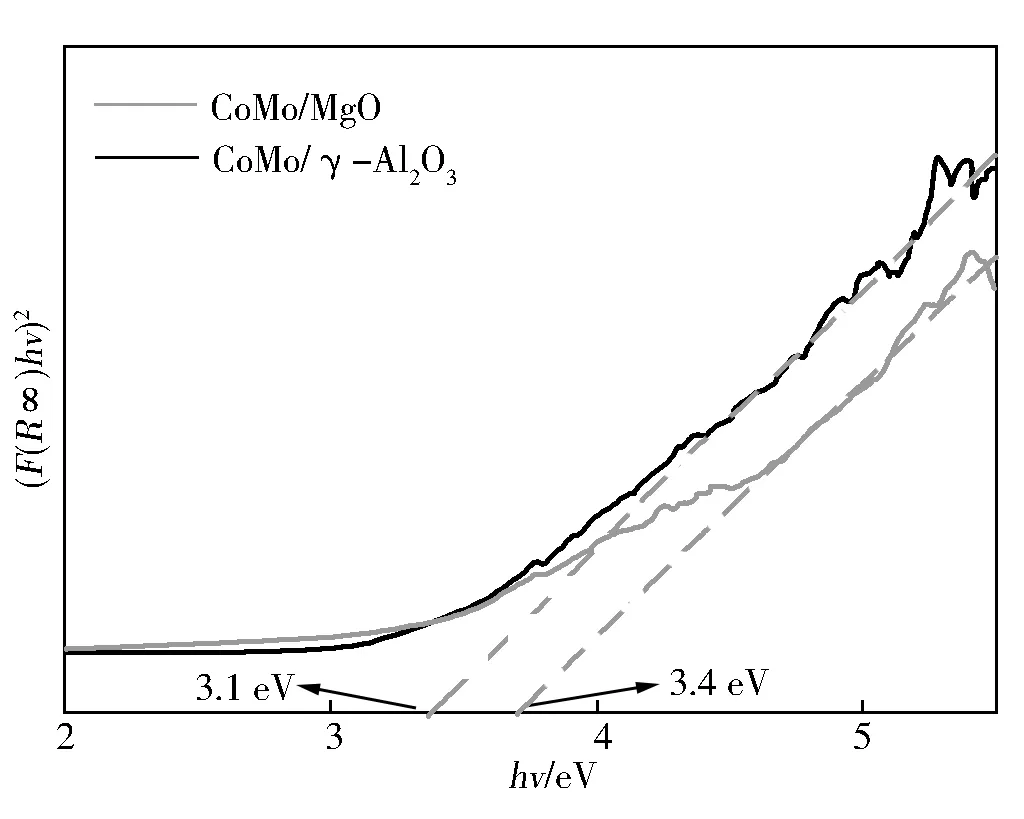
图4 CoMo/MgO、CoMo/γ-Al2O3催化剂煅烧后的(F(R∞)hv)2和能带能量关系
2.1.4 XPS表征
为了获得硫化催化剂中金属的状态,对CoMo/MgO和CoMo/γ-Al2O3硫化后的催化剂进行了XPS表征,结果见图5。

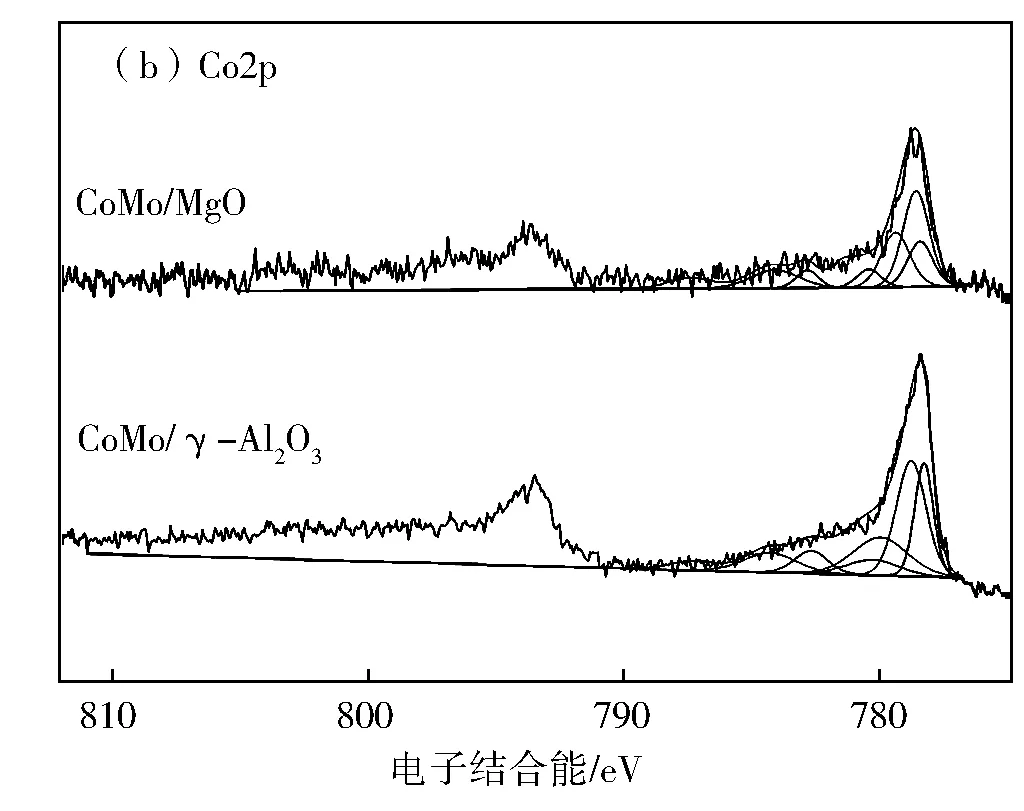
图5 硫化催化剂CoMo/MgO和CoMo/γ-Al2O3的XPS谱图及其对应分解峰
对于金属硫化物催化剂,表面Mo物种以MoS2、MoOxSy和MoO33种形式存在,对应的电子结合能分别是(229.0±0.3)eV (MoS2,Mo4+)、(230.5±0.2)eV (MoOxSy,Mo5+)和(232.2±0.2)eV(MoO3,Mo6+)[4]。因此,对CoMo/MgO和CoMo/γ-Al2O3催化剂上Mo物种的XPS光谱(图5(a))进行分峰拟合分析,并计算两个催化剂上不同Mo物种的相对含量,结果见表2。由表2可知,CoMo/MgO催化剂上Mo物种的硫化程度为69.8%,低于CoMo/γ-Al2O3催化剂(77.0%),说明Mo物种在MgO载体上还原温度更高,这是因为MgO的碱性位点比γ-Al2O3的多,加强了与酸性钼物种之间的相互作用,因此,CoMo/MgO催化剂上的钼氧化物可以更好的分散在MgO载体上,而且在CoMo/MgO催化剂上形成的多为八面体钼物种,通过硫化可以形成更多的MoS2活性相[11]。同时,两个催化剂上Co物种的XPS结果表明,均存在Co-(Ⅱ)、CoMoS和Co9S83种Co物种的相,分别对应的电子结合能为(781.1±0.5 eV)(Co-(Ⅱ))、(778.6±0.2 eV)(CoMoS)和(778.3±0.4 eV)(Co9S8)[4]。表3给出了不同Co物种在两个催化剂表面的含量。由表3结果可见,CoMo/MgO催化剂上Co物种参与形成CoMoS活性相的比例为58.9%,多于CoMo/γ-Al2O3催化剂(56.4%),说明在催化剂CoMo/MgO上形成了更多的CoMoS活性相。其中,CoMoS活性相的含量与催化剂的加氢脱硫性能密切相关。一般而言,CoMoS活性相的含量越高,催化剂的加氢脱硫性能越好。因此与CoMo/γ-Al2O3催化剂相比,CoMo/MgO催化剂的加氢性能更高。
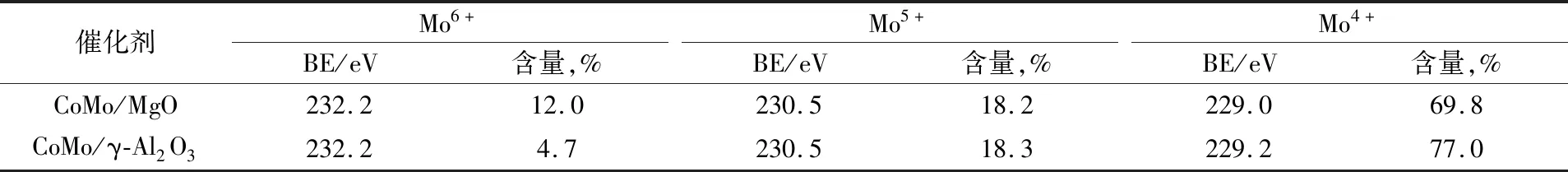
表2 硫化催化剂CoMo/MgO和CoMo/γ-Al2O3表面Mo3d物相XPS拟合结果

表3 硫化催化剂CoMo/MgO和CoMo/γ-Al2O3表面Co2p物相XPS拟合结果
2.1.5 TEM表征
图6为硫化后CoMo/MgO和CoMo/γ-Al2O3催化剂的TEM照片。从图6可见,在CoMo/MgO和CoMo/γ-Al2O3催化剂上均能明显的观察到MoS2相的晶格条纹。根据Topsøe等[1]提出的Co-Mo-S模型可知,Co-Mo-S活性相中存在Type Ⅰ和Type Ⅱ两种结构,其中Type Ⅱ型Co-Mo-S相的加氢脱硫活性远高于Type Ⅰ型Co-Mo-S相。对于CoMo/γ-Al2O3催化剂,在Type Ⅰ型结构中,还存在着大量的Mo-O-Al键,Mo难以完全硫化,从而不利于HDS活性的提高。分别在CoMo/MgO和CoMo/γ-Al2O3催化剂上的不同样品区域拍摄TEM照片,对每个催化剂上MoS2相的片层长度(L)和堆垛层数(N)进行统计分析,其结果见图7。从图7可知,CoMo/MgO和CoMo/γ-Al2O3催化剂上的MoS2活性相长度分布情况类似(图7(a)),主要集中在3~6 nm附近,但是CoMo/γ-Al2O3催化剂的长度分布比例较大;MoS2活性相在CoMo/MgO催化剂上的堆垛层数主要为1~2层(图7(b)),小于CoMo/γ-Al2O3催化剂上MoS2的堆垛层数(2~3层)。

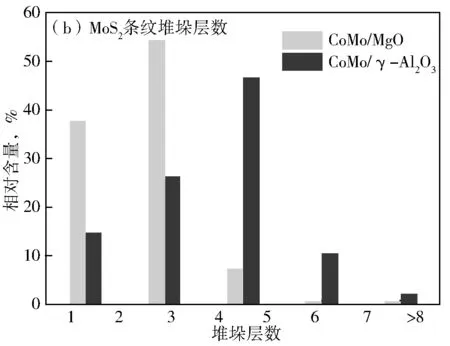
图7 硫化催化剂CoMo/MgO和CoMo/γ-Al2O3的MoS2晶体条纹分布

表4 硫化催化剂CoMo/MgO和CoMo/γ-Al2O3的MoS2晶体平均长度平均堆垛层数和分散度(DMo)
2.2 CoMo/MgO和CoMo/γ-Al2O3催化剂加氢脱硫活性评价结果
考察了CoMo/MgO和CoMo/γ-Al2O3催化剂对DBT加氢脱硫活性,结果见图8。由图8可知,在DBT加氢脱硫反应中,两个催化剂均呈现较好的稳定性,CoMo/MgO催化剂的催化性能明显优于CoMo/γ-Al2O3催化剂。例如,在反应进行20 h时,CoMo/MgO催化剂上DBT的转化率为88.3%,而CoMo/γ-Al2O3催化剂上的转化率仅为65.5%。因此,在DBT加氢脱硫反应中,与CoMo/γ-Al2O3催化剂相比,CoMo/MgO的加氢脱硫活性更高。
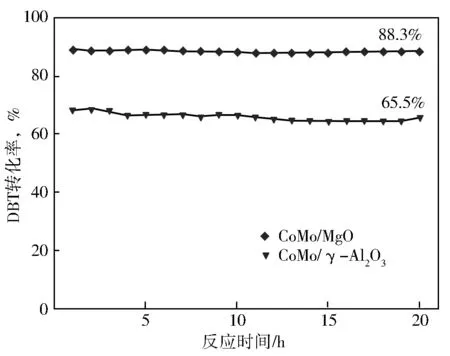
图8 CoMo/MgO、CoMo/γ-Al2O3催化剂上DBT加氢脱硫活性反应中转化率随着时间的变化曲线
在消除了传质对HDS反应的影响下,进一步比较了CoMo/MgO和CoMo/γ-Al2O3催化剂对DBT加氢脱硫本征活性,结果见图9。计算得到 CoMo/MgO催化剂的反应速率常数(kHDS)和催化转化频率(TOF)分别为0.268×10-6mol/(s·g)和3.4 h-1,而CoMo/γ-Al2O3催化剂的kHDS和TOF分别为0.207×10-6mol/(s·g)和2.8 h-1。显然,CoMo/MgO催化剂的本征加氢脱硫活性高于CoMo/γ-Al2O3催化剂。
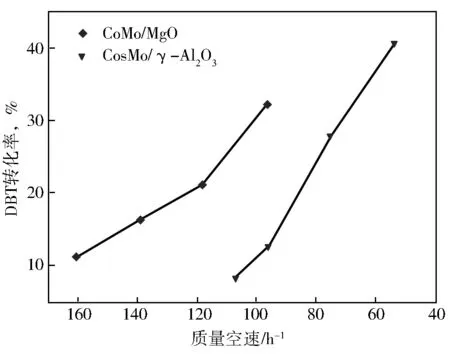
图9 CoMo/MgO、CoMo/γ-Al2O3催化剂上DBT的加氢脱硫活性反应中转化率(x)随质量空速的变化曲线
催化剂的加氢脱硫性能与金属硫化物的结构有关。与γ-Al2O3载体相比,MgO载体具有更多的碱性位点,从而有利于与酸性钼物种之间的相互作用,进而提高钼物种的分散性。通过UV-vis分析发现,在CoMo/γ-Al2O3催化剂上生成了CoAl2O4相,通过硫化,CoMo/MgO催化剂上有更多的Co参与形成CoMoS活性相。因此经XPS表征发现,在CoMo/MgO催化剂上CoMoS活性相的比例高于CoMo/γ-Al2O3催化剂。此外CoMo/MgO催化剂的介孔体积明显大于CoMo/γ-Al2O3催化剂,在反应过程中有利于大分子含硫化合物的扩散,使得含硫化合物更容易的发生吸附、催化反应,对催化性能的提高起到促进作用。因此,CoMo/MgO催化剂的加氢脱硫性能优于CoMo/γ-Al2O3催化剂。
3 结 论
以MgO为载体,制备了CoMo/MgO催化剂,在DBT的加氢脱硫反应中呈现了较好的催化性能,且其活性要优于CoMo/γ-Al2O3催化剂。CoMo/MgO催化剂具有高的加氢脱硫活性主要归功于MgO载体的碱性。有利于加强载体与酸性钼物种之间的相互作用,进而提高钼物种的分散性。此外,与CoMo/γ-Al2O3催化剂相比,在CoMo/MgO催化剂上生成了更多的八面体钼物种,通过硫化过程,可以形成更多的CoMoS活性相,从而促进DBT的加氢脱硫活性。
