儿童先天性肺气道畸形20 例临床分析
2020-02-19刘新锋刘晓娟赵志妙张中平
刘新锋 刘晓娟 赵志妙 张中平
河北医科大学附属河北省儿童医院(河北石家庄 050031)
儿童先天性肺气道畸形(congenital pulmonary airway malformation,CPAM)是一种少见的肺组织紊乱的错构瘤,伴有不同程度的囊性改变,以肺分支形成过程中气道模式异常为特征,由未成熟细支气管的异常分支形成[1-2],无明确病因或明确的遗传联系。在先天性肺部畸形中,胎儿CPAM的发病率约为25%[3],活产婴儿中的发病率为1/25000~1/35000,先天性肺囊性疾病约15%~50%为CPAM[4]。目前国内外对儿童CPAM的外科诊治及临床病理研究较多,而对于从内科方面着手的临床特点的综合研究鲜有报道。为此,本研究回顾分析20例CPAM患儿的临床资料,总结其临床特点。
1 临床资料
回顾分析2016 年1 月至2018 年12 月,在河北省儿童医院诊断为CPAM的20例患儿的临床资料。20例患儿中,男12 例、女8 例;平均年龄(30.4±7.6)月(1个月~8岁),其中1岁以下患儿3例,1~3岁患儿12例,3岁及以上患儿5例。
20例患儿中,临床表现为反复咳嗽、喘息,发热17例,胸闷1例,呼吸困难2例;其中误诊为支气管扩张1例,误诊为肺大疱及气胸1例。4例患儿为双侧病变;16例为单侧病变,其中累及左侧7例、右侧9例。2例患儿合并心脏畸形,1例为三房心、另1例为室间隔缺损。
胸部CT 表现:14 例表现为大囊型,均伴有纵隔移位;6例为小囊型(图1)。
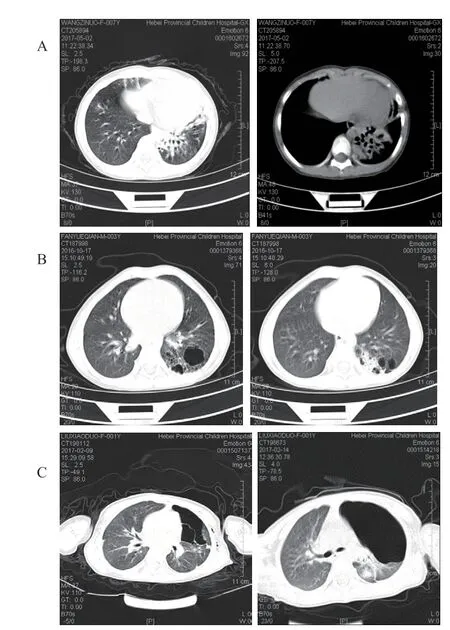
图1 CPAM 胸部CT 表现
20 例患儿均在肺部感染控制后进行肺叶或肺段切除术,手术全部获得成功。手术获取的肺组织标本采用苏木精-尹红(HE)染色,依据Stocker 分型进行病理分型。
20例患儿的病理分型:无0型和Ⅲ型,Ⅰ型14例,Ⅱ型5例,Ⅳ型1例。Ⅰ型镜检表现:大小不等的囊腔内衬假复层纤毛柱状上皮,部分呈乳头状突起,囊壁周围有平滑肌和弹力组织,周围肺组织出血,肺泡腔内淤血、水肿。Ⅱ型镜检表现:肺叶组织内见少量微囊,内衬单层纤毛矮柱状上皮或立方上皮,外围少量平滑肌,周围肺泡大片融合、扩张,内含血细胞及粉染液体,肺泡壁充血、出血,淋巴滤泡增多,边缘区肺泡组织实性变,呈小叶性肺炎样改变,肺膜充血、出血。Ⅳ型镜检表现:囊腔大,壁薄,内衬纤毛柱状上皮,纤维性囊壁内脉管增生、扩张。Ⅳ型患儿1例,表现为肺大泡和气胸,考虑与囊腔位于肺周边远侧肺泡,多个囊腔相通,肺泡易融合为肺大泡,且脏层胸膜破裂后易引起气胸。2例心脏畸形患儿均为Ⅱ型。见图2。
20 例患儿术后随访时间1~24 个月不等,患儿均恢复良好,复查胸部CT均无异常变化。
2 讨论
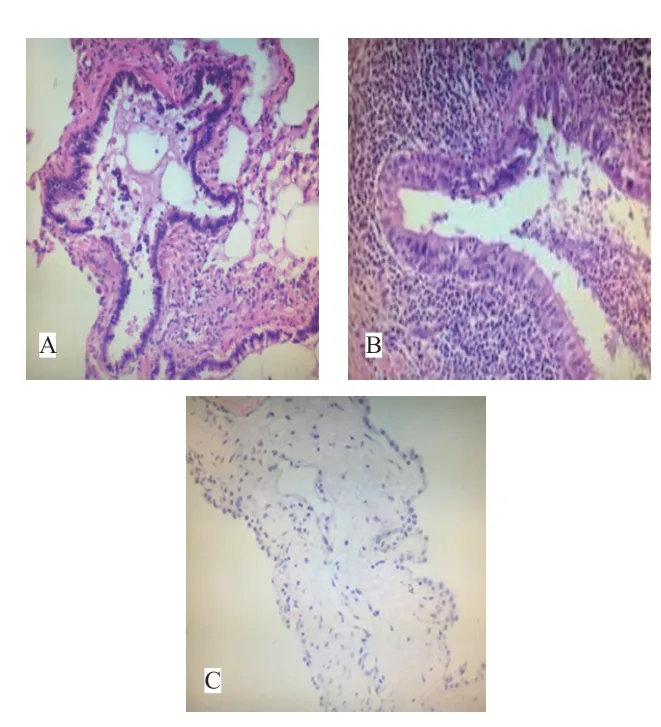
图2 CPAM 镜下病理(HE×100)
CPAM在1949年首先由Ch'in和Tang[5]将其作为一种少见的发生在未成熟胎儿或死产婴儿伴有全身水肿的病变提出,以前称先天性肺囊腺瘤样畸形。
国外研究认为,CPAM 的男女发病率无差别[1]。但国内研究认为,男性CPAM 的发病率高于女性[6]。本研究男性多于女性。因研究样本小,有待进一步证实。
CPAM 的发病机制尚不完全清楚。有研究者认为,CPAM发病机制是在肺形态发生过程中出现异常的气道模式和分支而导致肺囊肿的出现[7]。虽然确切的细胞参与机制尚不清楚,但认为许多潜在的基因与肺囊肿的形成有关。如有研究推测,部分由胶质细胞源性神经营养因子介导的HOXB5基因缺陷与一小部分CPAM 的发病机制有关[8]。甲状腺转录因子-1 在CPAM Ⅰ、Ⅱ、Ⅲ型中的表达有差异,可以作为CPAM组织分型的一个重要标记。而错配修复基因2(hMSH-2)在CPAM Ⅰ、Ⅱ、Ⅲ型中表达量均低于正常儿童肺组织,提示在CPAM中可能有hMSH-2基因转录水平的下降,从而影响hMSH-2 蛋白的表达,使之不能充分发挥修复错配DNA 的功能,产生DNA 复制错误,导致囊腔的形成[9]。国外研究发现,甲状腺转录因子1,纤维母细胞生长因子7,纤维母细胞生长因子9,纤维母细胞生长因子10,脂肪酸结合蛋白7,克拉拉细胞标记10,SOX2转录因子和改变整合蛋白胞质信号都可能在CPAM的发病机制中起到作用[10]。
新生儿期CPAM 常表现为不明原因的呼吸窘迫或无任何症状,婴幼儿及儿童常表现为反复呼吸道感染,成人罕见。CPAM可伴有各个系统的畸形(心血管、消化道、泌尿系统、神经系统)。本研究中患儿均有呼吸道感染症状,反复咳嗽、喘息、发热,胸闷,呼吸困难。少数被误诊为支气管扩张、肺大泡及气胸。
研究发现,CPAM多累及单侧单叶,上、下叶均可受累,以下叶多见,中叶少见,多叶或双侧累及极为罕见[11]。本研究中患儿单侧受累明显高于双侧,左右侧受累无显著差异。
CPAM 的影像学表现主要有3 种[12]:①大囊型,有两种表现形式,一种为肺野内巨大单发含气囊腔;另一种为大小不均的多个含气大囊。此型张力大,易致纵隔移位,常伴有纵隔肺疝。②小囊型,表现为多发不规则类圆形薄壁囊腔,类蜂窝状改变,伴感染时囊壁增厚。③实性型,表现为类似肿块、肺不张的改变。本研究患儿中,14例表现为大囊型且均伴有纵隔移位,6例为小囊型,未见实性型。
根据囊肿的大小和数量按病理学特点将CPAM分为5种类型。0型:最少见,主要邻近支气管树,常胎死宫内;Ⅰ型:最常见,多累及单个肺叶;Ⅱ型:通常仅累及一个肺叶,60%的病例伴有其他异常,包括心脏畸形、肾发育不全、胃肠闭锁、骨骼异常;Ⅲ型:并发畸形较多,常累及整叶或一侧肺,较易出现胎儿水肿,往往胎死于宫内、预后差;Ⅳ型:少见,位于肺的外围部,多个囊腔相通,但与支气管不相通。本研究患儿中,最多为Ⅰ型14例,Ⅱ型5例,Ⅳ型1例,未见0型和Ⅲ型。
由于恶性转化和反复呼吸道感染的风险,手术仍是有症状CPMA 的基础治疗,但无症状CPAM 的手术治疗仍存在争议。有研究者认为,随着时间的推移,感染的比例越来越高,经过长时间的演变,手术会变得越来越困难,同时也有恶性肿瘤的风险。无症状CPMA手术切除与症状发生后的干预相比更有益,并发症更少[4]。但也有研究者认为,虽然CPAM 有发生恶性肿瘤的风险,但肿瘤的发生率是未知的,建议在了解可能的并发症后,密切观察是否有手术的必要。此外,预防性切除CPAM病变并不总是完全具有保护作用,即使在切除病灶后,仍有发生恶化的可能[13]。本研究患儿均有呼吸道感染症状,均进行了手术切除病变组织,术后随访时间1~24个月不等,均恢复良好,复查胸部CT均无异常变化。
