H2O2活化活性炭对U(VI)的吸附研究
2020-01-17魏青峰石艳明李同同庹先国王彦惠
魏青峰 石艳明 李同同 庹先国 王彦惠
1(西南科技大学国防科技学院 绵阳 621010)
2(中广核工程有限公司 深圳 518000)
3(四川轻化工大学 自贡 643033)
在核能积极开发利用的今天,大量高毒性的放射性核素也在侵入环境,造成的水污染日益严重。因在矿石的开采、燃料加工和后处理等任何环节都可能泄露,铀被认为是最严重的水污染物之一[1-2]。六价铀(U(VI)),在水中具有较高的溶解度和迁移率,因而成为主要研究对象[3]。在众多水污染处理方法中,吸附法是最为成熟的一种。成本低、效率高、操作简易等优点使其被广泛运用于放射性水污染处理当中。其中活性炭是处理环境中有毒有害物质的常见吸附剂。普通的商品活性炭对U(VI)的吸附性能不强,存在吸附容量低、平衡吸附时间长等缺陷,因此本研究利用过氧化氢(H2O2)对活性炭进行氧化活化,表征了活化前后活性炭的表面与结构性质,探究过氧化氢(H2O2)增强活性炭对U(VI)吸附性能的机理;考察了活性炭对溶液中U(VI)吸附性能的影响因素,为处理铀污染水溶液的吸附剂的发展提供参考。
1 实验与方法
1.1 实验原料
活性炭颗粒(成都金山化学试剂有限公司);过氧化氢(H2O2,30%),盐酸(HCl,37%)和硝酸(HNO3,68%);偶氮胂III(上海阿拉丁生物化工技术有 限公司);标 准 溶液 U(VI)(pH=5,浓度为200 mg·L-1硝酸铀酰);全程使用去离子水。
1.2 活性炭的活化
用超纯水将活性炭颗粒浸泡1 h后再冲洗3次,然后放置100℃烘箱内烘6 h。将烘干后的活性炭研磨至0.075~0.150 mm,并再次放入100℃烘箱内烘干至质量恒定。
取10 g处理后的活性炭分别加入500 mL的体积分数分别为15%、30%的H2O2溶液中,并置于超声水浴器中3 h后离心,将活性炭在70℃下干燥12 h,得到的活化活性炭分别记为15%-AC、30%-AC。
1.3 活化前后活性炭的表征
用傅立叶变换红外光谱(Fourier Transform Infrared spectroscopy,FT-IR)测定表面的性质(光谱范围为 4 000~400 cm-1);用梅特勒-托利多公司TGA/SDTA851e型热重/热差分析仪进行了热重分析 热 重 分 析(Thermogravimetric Analysis,TGA)(50 mL·min-1的N2为载气,10 ℃·min-1的升温速率,温度为30~1 000℃);用日立4700型扫描电子显微镜(Scanning Electron Microscope,SEM)进行了扫描电镜观察;用康塔仪器公司的比表面和孔隙分析仪(NOVA3000)测定比表面积与孔径分布(250℃下真空脱气12 h)。
1.4 吸附实验
将0.05 g活性炭置于离心管中,加入7 mL去离子水,恒温振振荡24 h后待活性炭间隙中的空气被完全排除后,再加入2 mL U(VI)溶液摇晃混合。调节pH到设定值之后定容,继续振荡,到达吸附设定时间后离心20 min以实现固液分离。最后,取1 mL的上清液采用分光光度法对U(Ⅵ)进行测定。通过式(1)计算U(VI)在活性炭上的吸附率η(%)[4]:

式中:Caq是 U(VI)浓度,g·L-1;C0是 U(VI)的初始浓度,g·L-1。
2 结果与讨论
2.1 活性炭的表征
2.1.1 FT-IR表征与分析
活化前后与吸附前后的活性炭表面官能团的FT-IR光谱分析如图1所示。对比活化前后的光谱,2 920 cm-1、2 850 cm-1对应于芳香族烯烃基团中CH的伸缩振动;1 452 cm-1对应于芳香族中C=C的伸缩振动,这些峰的减弱证明活化过程破坏了芳香族基团分子链,并伴随有更多中孔的形成[5]。3 430 cm-1(羟基-OH伸缩振动)的峰是随着H2O2活化强度而增强的,说明活化后-OH增多[6]。对比吸附前后的光谱,最明显的是3 430 cm-1、2 920 cm-1、2 850 cm-1、1 640 cm-1(C=O)、1 452 cm-1(C=C)等吸收峰的减弱或消失,这表明U(Ⅵ)离子与活性炭表面的-OH、C=O、C=C等官能团产生络合效应,从而形成有效的吸附行为[7]。
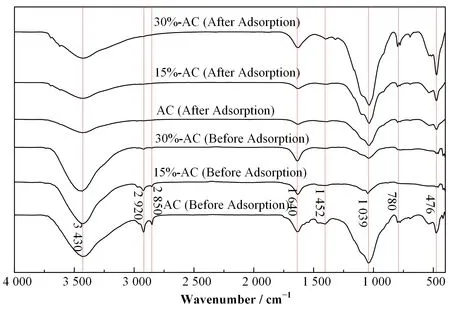
图1 活化前后与吸附前后的FT-IR光谱Fig.1 FT-IR spectra ofAC before and after modification and adsorption
2.1.2 TGA表征与分析
活化前后活性炭的热重曲线(Thermogravimetric,TG)如图2所示。曲线显示,活化前后活性炭的热重曲线形成明显的三个阶段。三种活性炭在25~190℃之间形成相近的重量损失,这是由于其自身水分在高温下挥发造成的[8];据报道,在190~700℃的重量损失只要是活性炭表面的氢氧基团(如:羟基)的热解所形成的[9];而在700℃以上所形成的重量损失这对应的是碳氧基团(如:羰基、羧基、酯基)的热解[10]。在高温分解阶段,随着活化的H2O2浓度增加,对应的氢氧、碳氧基团的热解失重明显增加。这说明H2O2浓度越高,在活性炭表面形成的碳氧基团含量越高。
FT-IR与TGA结果说明活化过程在活性炭表面引入了大量的官能团,活化过程示意如图3所示。这些官能团能够形成更多吸附位点,这提升了吸附性能。

图2 活化前后活性炭的TG曲线Fig.2 TG curve ofAC before and after activation

图3 活化过程Fig.3 Schematic diagram of activation process
2.1.3 BET表征与分析
使用氮吸附/解吸等温线的Brunauer-Emmett-Teller(BET)方法对活化前后活性炭的微观结构进行表征,结果如表1所示。
由表1可以看出,活化后活性炭的比表面积略微下降,这可能是H2O2的强氧化性和酸性腐蚀活性炭基础结构,引起了活性炭某些结构坍塌[11];但活化后活性炭的总孔容、中孔孔容和平均孔径均有明显的增大,这是吸附容量提升的重要原因之一[12]。同时发现相比于15%-AC,30%-AC的各项孔隙结构反而有所减少,所以过强的活化浓度不一定对活性炭性能的提升有利。

表1 活性炭活化前后BET孔径分析结果Table 1 BET analysis ofAC before and after activation
2.1.4 SEM表征与分析
扫描电子显微镜(ScanningElectron Microscope,SEM)能够更加直观地观察活化前后活性炭的表面形态。图4显示了在相同放大倍数下(Mag=5.00 K X),AC、15%-AC和30%-AC的SEM显微照片,结果证实了H2O2活化后的活性炭在孔隙数量上有明显变化。随着活化的进行,15%-AC和30%-AC出现更多的孔隙,这对吸附性能的提升是有利的;同时也发现,相比于30%-AC,15%-AC的孔隙结构与数量更加明显清晰,这是因为高浓度的H2O2对AC的酸化、氧化行为会腐蚀破坏活性炭表面,导致某些结构坍塌,反而降低了吸附性能[13]。

图4 活化前后活性炭SEM图Fig.4 SEM charts ofAC before and after activation
2.2 接触时间对吸附的影响
通过改变活性炭与U(Ⅵ)离子溶液的接触时间,研究了活化前后的活性炭的吸附性能。图5为不同接触时间下,U(Ⅵ)离子在AC、15%-AC和30%-AC上的吸附率。由图4可知,活性炭对U(Ⅵ)的吸附可以分为0~50 min、50~90 min和大于90 min三个阶段。0~50 min阶段形成快速吸附行为,这是因为吸附初期,活性炭表面吸附位点处于等待状态,表面接触的U(Ⅵ)能够被及时吸附;50~90 min阶段的吸附效率减缓,这是因为该阶段活性炭吸附以物理吸附和静电吸附结合的方式进行,U(Ⅵ)离子在固液表面的扩散与静电吸引需要一定时间完成。在90 min以后,吸附率非常缓慢地增加,这是因为当吸附位点被基本占据后,吸附行为进入平衡阶段,溶液中游离的U(Ⅵ)离子被吸附的概率变得非常小,只能随着时间的增加而缓慢吸附。对比AC与15%-AC,在相同条件下,吸附容量增加了68%。这是证明了利用H2O2活化能够提升活性炭对U(Ⅵ)的吸附容量。

图5 不同接触时间对吸附率的影响Fig.5 Effect of different contact time on adsorption rate
为了更清晰地阐明吸附行为,利用实验数据拟合了动力学线性模型,发现吸附更加符合准二阶吸附模型:

式中:Qe为理论平衡吸附容量,mg·g-1;Qt(mg·g-1)为t(min)时刻的吸附容量;k2为准二级吸附速率常数,g·mg-1·min-1。方程拟合结果示于图 6中,相关参数列于表2中。
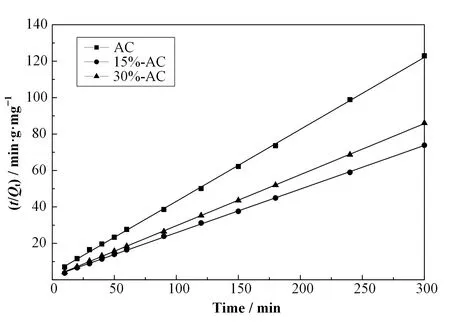
图6 准二级动力学拟合Fig.6 Pseudo-second-order dynamics fitting
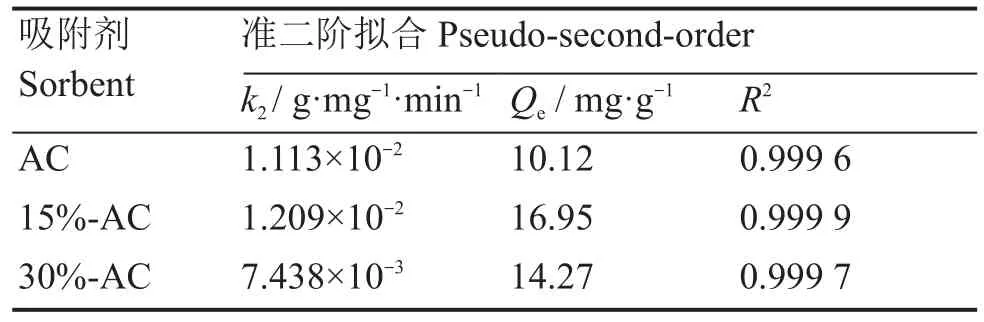
表2 准二级动力学拟合参数Table 2 Pseudo-second-order kinetic fitting parameters
从表2可以看出,三种活性炭的均有很好的准二级动力学线性拟合(R2>0.999);且AC、15%-AC和30%-AC 的 实 验 平 衡 吸 附 量(9.60 mg·g-1、16.20 mg·g-1、13.8 mg·g-1)是非常接近理论平衡吸附量(10.12 mg·g-1、16.68 mg·g-1、14.28 mg·g-1);这说明活化前后的活性炭对U(Ⅵ)的吸附过程并非只有物理吸附,而是个多基元反应组成的复杂行为[14]。
2.3 溶液pH的影响
溶液的pH对活性炭吸附U(Ⅵ)有着显著的影响,吸附结果示于图7。在pH<5时,随着pH的增加,U(VI)在AC、15%-AC和30%-AC上的平衡吸附率逐渐增加到各自的最大值(50.62%,78.72%,69.85%);在pH>5时,平衡吸附率随pH的增大而缓慢下降。这是因为是pH会影响活性炭表面的吸附位点和U(Ⅵ)在溶液中的形态[15]。据报道,活性炭表面具有大量的负电荷吸附点[16],能够吸引周围带正电荷的 U(Ⅵ)(如:UO2OH+、UO22+等);但在强酸环境下,库仑力先拉动具有高的电荷质比的H+,减弱了与U(Ⅵ)离子的结合;而在碱性环境下,U(Ⅵ)离子与OH-络合,减少U(Ⅵ)离子表面正电荷数量并增大了U(Ⅵ)的粒子半径(如:UO2(OH)3-、(UO2)3(OH)7-),导致吸附率下降[17]。因此活化后的活性炭吸附U(Ⅵ)离子的最佳pH为5。

图7 不同pH对吸附率的影响Fig.7 Effect of different pH values on adsorption rate
2.4 固液比的影响
吸附介质与溶液的固液比对U(Ⅵ)吸附率的影响示于图 8中。当固液比为 1~15 g·L-1,AC 对U(Ⅵ)吸附率逐渐增长且未出现减缓趋势;当固液比<8 g·L-1时,15%-AC的吸附率处于快速上升阶段,之后趋于平缓。这是因为活化后的活性炭因具有更佳的吸附容量,当活性炭用量过大时,部分吸附位点处于空置状态,导致活性炭的利用率较低。还可以看出,要达到相同的效果,15%-AC的固液比小于30%-AC,说明前者的吸附容量大于后者,这差异说明使用过量的H2O2对活化并不有利,这一结果与前文的表征现象也是相互对应的。活化后的活性炭吸附U(Ⅵ)离子的最佳固液比为8 g·L-1。

图8 不同固液比对吸附率的影响Fig.8 Effect of different solid-to-liquid ratios on adsorption rate
2.5 初始浓度的影响

图9 不同U(Ⅵ)初始浓度对吸附容量的影响Fig.9 Effect of initial concentration of U(VI)on adsorption rate
图9 反映了活化前后活性炭对U(Ⅵ)吸附容量与U(Ⅵ)初始浓度的关系。如图9所示,当溶液中U(Ⅵ)的初始浓度在10~60 mg·L-1时,活化前后的活性炭吸附容量随着初始浓度的增加而逐渐上升,三者相差不大,这是因为在溶液中的U(Ⅵ)离子量很少的情况下,活性炭吸附位点是充足的,接近活性炭表面的U(Ⅵ)离子都能够被及时的吸附。然而,当溶液中U(Ⅵ)的初始浓度>60 mg·L-1时,AC开始进入饱和吸附量阶段,吸附位点被大部分占据,浓度的增加也无法提升U(Ⅵ)离子被吸附几率,不能再形成有效的吸附行为;而15%-AC和30%-AC的吸附容量则继续上升,这证明H2O2活化过后的活性炭具有更高的吸附容量。活化后的活性炭吸附U(Ⅵ)离子的最佳初始浓度为80 mg·L-1。
为了更好地研究活性炭与U(Ⅵ)之间的相互作用吸附模型,将初始浓度对吸附影响的实验数据拟合到两种常见的等温模型中:
Langmuir等温吸附方程:

Freundlich等温吸附方程:
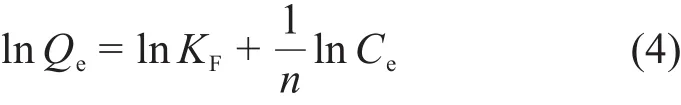
式中:Qe为平衡吸附状态下活性炭的吸附容量,mg·g-1;Ce为平衡吸附状态下溶液中 U(Ⅵ)浓度,mg·L-1;KL为Langmuir吸附常数;KF为Freundich吸附常数;1/n,吸附强度常数。相关数据列入表3中。
从表3可以看出,对比两种模型的拟合相关系数,活化前后的活性炭对U(Ⅵ)的吸附过程在Langmuir等温吸附模型上具有更高的拟合相关系数(R2>0.99)。这表明活化后的15%-AC与30%-AC的表面在性质上是均匀的[16]。

表3 等温吸附模型拟合参数Table 3 Langmuir and Freundlich parameters U(VI)adsorption
2.6 温度的影响
通过设置不同吸附温度,研究了温度对活化活性炭吸附U(Ⅵ)离子的影响,结果示于图10。结果表明在25~55°C的范围内,吸附率呈现先增长再下降的趋势,在35°C时,出现最佳的吸附效果,这是因为高温对离子游离程度和含氧基团活性具有一定的影响;但吸附行为在此范围内受到温度的影响不明显,这说明活化活性炭能够在较宽的温度范围内有效吸附溶液中的U(Ⅵ)离子。

图10 不同温度对吸附率的影响Fig.10 Effect of different temperature on adsorption rate
2.7 离子种类的影响
U(Ⅵ)离子的去除和活性炭的吸附性能受到溶液中其他共存离子的影响。因此选择一些阴阳离子,研究了不同离子种类对15%-AC吸附U(Ⅵ)离子的影响。每种离子的浓度保持在8.5×10-5mol·L-1,与之前实验中 U(Ⅵ)离子浓度相同。结果示于图11。
研究发现不同种类的离子对15%-AC吸附U(Ⅵ)离子产生不同的趋势。如图11(a)所示,在阳离子共存条件下,Na+、K+、Mg2+和 Ca2+均能抑制吸附,抑制强度为Ca2+>Mg2+> K+> Na+。这是因为与低价阳离子相比,高价阳离子与15%-AC表面带了更多的负电荷,更容易与15%-AC表面的活性官能团竞争并结合[18]。另外,在相同的价态条件下,Ca2+的水化半径小于Mg2+,K+的水化半径小于Na+。根据竞争吸附规律,水合离子半径越小,吸附竞争力越强。如图11(b)所示,在阴离子共存条件下,Cl-和对U(VI)的吸附影响不大。其它阴离子对U(Ⅵ)的吸附有抑制作用,抑制强度为。研究表明,在水溶液中易于结合形成多核配合物,降低了U(Ⅵ)离子与表面官能团结合能力,增加了15%-AC吸附U(VI)的难度。
2.8 循环吸附性能
吸附剂的重复利用具有经济效益,是评价其性能是否优异的重要指标。利用15%-AC在最佳的吸附条件下吸附U(Ⅵ)离子后,使用浓度为1 mol·L-1的HCl溶液解吸15%-AC上的U(Ⅵ),将解吸后的15%-AC在相同条件下再次吸附U(Ⅵ)离子,吸附/解吸6次[19],结果示于图12。

图11 阳离子(a)及阴离子(b)对U(Ⅵ)在15%-AC上吸附的影响Fig.11 Effect of cations(a)and anions(b)on the adsorption ratio of U(Ⅵ)on 15%-AC

图12 循环吸附性能Fig.12 Cyclic adsorption performance
结果显示:吸附/解吸过程对15%-AC有一定的影响,在经历过6个循环后使其吸附性能下降了20%,这是因为15%-AC与AC最大的区别在于活化后的活性炭表面具有更多含氧基团与孔隙结构,但在用HCl溶液解吸的过程中会极大地破坏这些含氧基团;同时发现在多次循环吸附后15%-AC的性能始终由于AC,这表明活化形成的孔隙结构不会吸附/解吸过程破坏,能够稳定存在。这证明15%-AC能够多次利用,具有良好的经济效益。
3 结语
本文以活性炭作为基础吸附介质,利用15%和30%两种浓度的H2O2对AC进行活化,并结合仪器表征分析与批量静态吸附实验,研究了活化前后的活性炭对溶液中U(Ⅵ)离子的吸附性能。得出以下结论:
1)仪器表征证明,经H2O2活化后的活性炭,其表面增加了大量的氧化基团;比表面积虽小幅度下降,但孔隙、孔容、孔径均得到明显提升;改性后的活性炭具有更利于吸附的表面与结构性质。
2)活化过程中过量的H2O2浓度并不能形成更强的活化效果。批量静态实验数据表明,对U(Ⅵ)的吸附性能大小顺序为15%-AC>30%-AC>AC,且吸附行为符合准二级吸附模型和Langmuir等温吸附模型。在接触时间为150 min、pH为5.0、U(Ⅵ)初始浓度为 80 mg·L-1、固液比为 7.5 g·L-1、温度 为35°C等最佳吸附条件下,15%-AC吸附容量(16.20 mg·g-1)是AC吸附容量(9.60 mg·g-1)的 0.68倍。研究证明了,过氧化氢活化行为能增强活性炭对U(Ⅵ)吸附性能。
3)常见的共存阴阳离子会对吸附行为产生抑制效果,阳离子抑制强度为Ca2+>Mg2+> K+> Na+,阴离子抑制强度为CO32->HCO3->SO42-。吸附/解吸过程会一定程度影响吸附性能,但多次循环吸附后的15%-AC依旧优于AC。
