肺癌特效药艾维替尼的合成工艺研究
2020-01-14林韦康江晓明武立靖
林韦康,高 奇,江晓明*,武立靖
(1.浙江云涛生物技术股份有限公司,浙江 绍兴 312369;2.艾依诺科技有限公司,浙江 诸暨 311800;3.南京康瑞医药化工有限公司,江苏 南京 210000)
0 引言
2019 年1 月,国家癌症中心发布的最新癌症统计数据显示,我国癌症发病率和死亡率均为全球第一,其中肺癌、结直肠癌、肝癌及女性乳腺癌等依然是我国主要的恶性肿瘤,按发病人数顺位排序,肺癌位居我国恶性肿瘤发病首位[1-2]。肺癌往往存在表皮生长因子受体(Epidermal Growth Factor Receptor,简称EGFR 或ErbB-1 或HER1)的高表达或异常表达,故治疗肺癌的常规手段是通过EGFR 酪氨酸激酶抑制剂(EGFR-TKI)来抑制EGFR 的高表达或异常表达。目前,国内市场上的EGFR-TKI 主要分为四代:一代代表药物为埃克替尼[3]、吉非替尼[4]和厄洛替尼[5],二代代表药物为阿法替尼[6],三代代表药物为奥希替尼[7]和本文将重点研究的艾维替尼[8]等,四代代表药物为EAI045[9],它们的化学结构式如图1 所示。众所周知,肺癌脑转移一直是医学界的一大难题,而普通的化疗药物较难穿透血脑屏障,通常要借助不良反应率高的放疗技术来治疗,随着靶向治疗即EGFR-TKI 的出现,肺癌脑转移患者有了新的治疗选择。采用一代EGFR-TKI 治疗肺癌的患者会在半年到一年左右出现耐药[10-11];与一代EGFRTKI 相比,二代EGFR-TKI 虽然作用靶点多,但不良反应强,患者耐受性差[12];三代奥希替尼可以更好地透过血脑屏障,目前临床上三代奥希替尼是首选药物,但奥希替尼价格太昂贵,国内每盒售价高达5.1 万元,很容易导致因病致贫的社会问题;四代EAI045 目前还停留在前期临床阶段。由艾森医药自主研发的三代艾维替尼马来酸盐若能顺利成为首个上市的国产三代EGFR-TKI,凭借价格优势,市场潜力将空前巨大。
艾维替尼(Avitinib,AC0010),化学名称为N-(3-{2-[3-氟-4-(4-甲基哌嗪-1-基)-苯基氨基]-7H 吡咯[2,3-D]嘧啶-4-酰氧基}-苯基)-丙烯酰胺,CAS:1557267-42-1,分子式:C26H26FN7O2,分子量:487.53,通常将艾维替尼制成药学上可接受的盐如艾维替尼马来酸盐、艾维替尼甲磺酸盐、艾维替尼酒石酸盐等制剂,供肺癌患者口服治疗。文献报道[13]艾维替尼的马来酸盐或盐酸盐相比于游离碱艾维替尼本身有三倍的生物利用度。艾维替尼对T790M 阳性患者有良好的耐受性和有效性,虽然穿透血脑屏障的能力较弱,但对无症状脑转移瘤有很好的控制作用[14],未来艾维替尼的规模化生产将是我国创新药物大力发展前所未有的机遇。
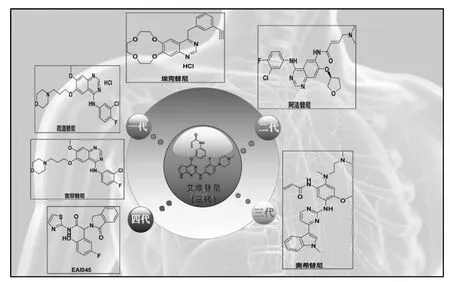
图1 肺癌代表性药物化学结构式Fig.1 The chemical structural formula of representative drugs for lung cancer
目前,国内外关于艾维替尼合成工艺的研究较少,本文在美国ACEA BIOSCIENCES 公司的发明专利US 20150210702[15]基础上进行改进、优化。该专利以2,4-二氯-7H-吡咯并[2,3-d]嘧啶为起始原料,用2-(三甲基硅烷基)乙氧甲基氯进行硅醚化后先与间硝基苯胺构建醚键,再与3-氟-4-甲基哌嗪苯胺发生Buchwald-Hartwig 交叉偶联反应构造亚胺键,最后经硝基还原、丙烯酰氨化、脱硅醚保护、成马来酸盐等反应,得到目标化合物,步骤较长,所用溶剂种类多,总收率偏低,特别是应该接近定量转化的硝基还原收率为85%,丙烯酰氨化收率为75%,脱硅醚保护需分两步处理,收率也只有65%。另外,几乎每步反应都要通过柱层析纯化,合成路线见图2。
为开发一套成本低、安全环保可控的艾维替尼合成新工艺,本文仍以价廉易得的2,4-二氯-7H-吡咯并[2,3-d]嘧啶为起始原料,先用乙酰氯保护起始原料吡咯环上的亚胺键,然后与间硝基苯酚钠反应成醚,接着与3-氟-4-甲基哌嗪苯胺进行Buchwald-Hartwig 交叉偶联成亚胺,再于冰乙酸中用还原铁粉还原硝基及脱乙酰基保护,最后伯胺上丙烯酰基得到目标产物。此外,也尝试将所得目标产物制成马来酸盐,结果良好。
1 实验部分
1.1 主要试剂与仪器
使用的主要试剂与仪器分别见表1 和表2。
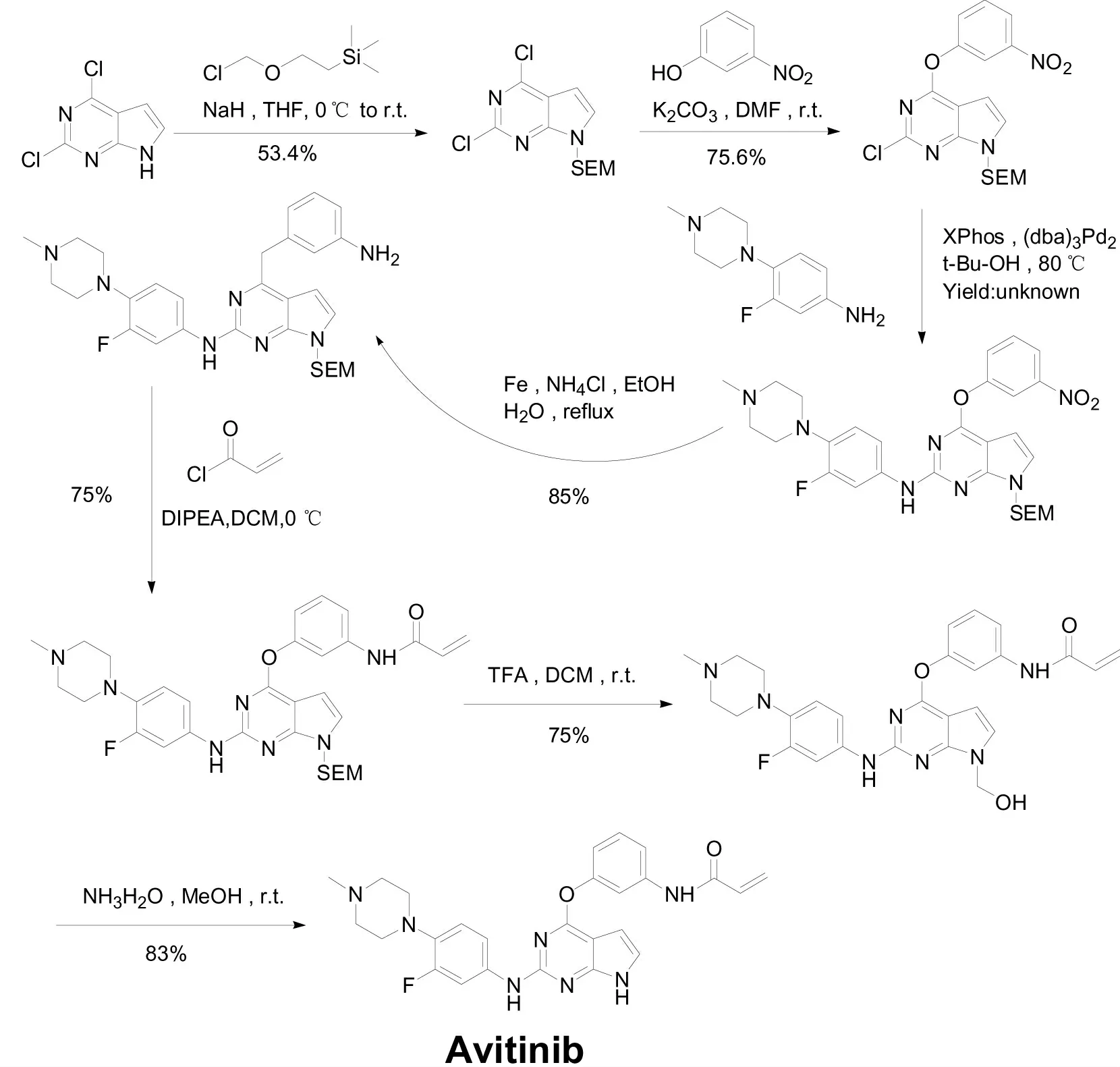
图2 专利US 20150210702 中的艾维替尼合成路线图Fig.2 The ROS of Avitinib in US 20150210702

表1 使用的主要试剂Tab.1 Main reagents

表2 使用的主要仪器Tab.2 Main instruments
1.2 合成方法
1.2.1 合成路线图
本文改进、优化后的艾维替尼合成路线图见图3。
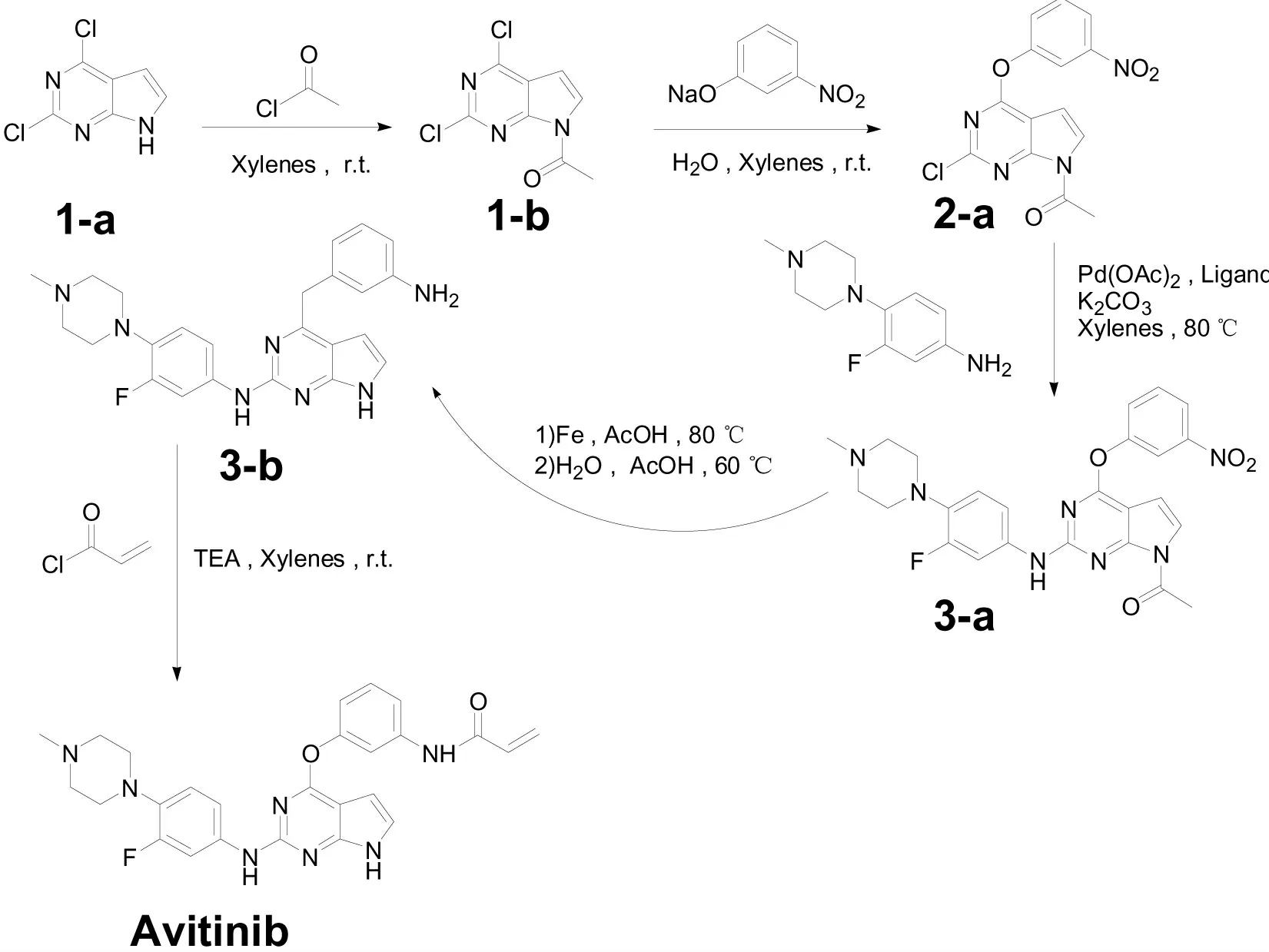
图3 艾维替尼的合成路线图Fig.3 The ROS of Avitinib
1.2.2 上保护(产物:1-b)
将二甲苯(300 mL)和2,4-二氯-7H-吡咯并[2,3-d]嘧啶(50.00 g,0.26 mol)投入500 mL 带有机械搅拌的四口瓶中,开启搅拌。控温≤25 ℃滴加乙酰氯(10.38 g,0.13 mol),滴毕,保温搅拌1 h。保温搅拌毕,抽滤,滤饼用适量二甲苯洗涤,洗涤液与滤液合并,为1-b 的二甲苯溶液,用适量饱和氯化铵溶液洗涤后去下一步工序。此处的滤饼为2,4-二氯-7H-吡咯并[2,3-d]嘧啶盐酸盐,将在2.1 章节叙述其处理方法。
1.2.3 醚化(产物:2-a)
将间硝基苯酚(18.00 g,0.13 mol)与水(100 mL)投入1000 mL 带有机械搅拌的四口瓶中,用32%氢氧化钠调pH=7.5,此时反应体系呈淡黄色澄清液体。随后加入适量相转移催化剂和1.2.2 中1-b 的二甲苯溶液。控温25 ℃~30 ℃搅拌12 h 后静置分层,分去下层水层,上层有机层用饱和食盐水洗涤后,加入适量活性炭和无水硫酸钠,搅拌30 min 后抽滤,得到黄色澄清液体,为2-a 的二甲苯溶液,进入下一步工序。
1.2.4 Buchwald-Hartwig 交叉偶联(产物:3-a)
将上一步工序所得2-a 的二甲苯溶液转移到500 mL 带有机械搅拌的四口瓶中,加入0.1 eq乙酸钯(II)及0.2 eq 配体、碳酸钾(41 g,0.30 mol),搅拌升温至80 ℃并保温5 h。保温毕,加入适量硅藻土和活性炭,搅拌30 min 后抽滤,滤饼用适量二甲苯洗涤,洗涤液与滤液合并,用饱和食盐水洗涤并用无水硫酸钠干燥后,油泵减压浓缩至瓶壁开始有料析出时停止浓缩,搅拌降温至10 ℃以下,抽滤,滤饼用适量二甲苯洗涤,得到黄色细小颗粒状固体湿品,为3-a,于80 ℃下真空干燥得干品56.01 g,以起始原料1-a 计,摩尔收率为85.23%。滤液过硅胶柱还可回收部分3-a,此处不作过多描述。
1.2.5 硝基还原和脱保护(产物:3-b)
将冰乙酸(200 mL)与3-a(10.11 g,0.02 mol)投入带有机械搅拌的500 mL 四口烧瓶中,搅拌溶清后一次性加入还原铁粉(4.50 g,0.08 mol),搅拌升温至80 ℃并保温3 h,保温毕,搅拌降温至30 ℃以下,滤除铁渣,铁渣用适量冰乙酸洗涤,洗涤液与滤液合并得到未脱保护的3-b 的冰乙酸溶液,铁渣去向将在2.4 章节讨论。
将得到的未脱保护的3-b 的冰乙酸溶液投入500 mL 带有机械搅拌的四口瓶中,加入30%盐酸(50 mL),搅拌升温至60 ℃并保温5 h,保温毕,搅拌降温至30 ℃以下,先用二甲苯(200 mL)萃取脂溶性杂质,再将萃取后的水层用32%氢氧化钠调节pH=9,抽滤,滤饼用适量自来水洗涤,于80 ℃下真空干燥,得到淡黄色粉末状固体干品8.39 g,为3-b,以3-a 计,摩尔收率为97.22%。此处的3-b 滤液去向将在2.4 章节讨论。
1.2.6 烯丙基化(产物:Avitinib)
将二甲苯(100 mL)和三乙胺(1.08 g,0.01 mol)、3-b(4.35 g,0.01 mol)投入250 mL 带有机械搅拌的四口瓶中,搅拌均匀,反应体系呈淡黄色澄清液体。控温25 ℃以下滴加丙烯酰氯(0.91 g,0.01 mol),滴毕,控温20 ℃~25 ℃,保温搅拌1 h,保温毕,抽滤,滤饼为三乙胺盐酸盐,滤液为Avi tinib 的二甲苯溶液,滤液用饱和食盐水洗涤后加入适量无水硫酸钠和活性炭,搅拌30 min,抽滤,滤液过快速硅胶柱后减压浓缩至干,得到淡黄色粉末状固体4.78 g,为Avitinib,以3-b 计,摩尔收率为97.26%,液相归一法含量为99.13%。实验结果说明,丙烯酰氯优先与伯胺反应,3-b 分子上两个仲胺键在反应体系浓度较低的情况下只会很少量地参与竞争反应,副反应产物可用冰乙酸/浓盐酸的处理方法返回3-b。
1H NMR(400 MHz,DMSO-d6):δ11.63(s,1H),10.33(s,1H),9.18(s,1H),7.68(s,1H),7.59(d,J=8.1Hz,2H),7.46~7.39(m,2H),7.10(t,J=4.0Hz,1H),7.02(d,J=8.1Hz,1H),6.82(t,J=8.1Hz,1H),6.47~6.38(m,1H),6.28(d,J=4.0Hz,2H),5.79(d,J=12.1Hz,1H),2.89(t,J=8.1Hz,4H),2.46(t,J=8.1Hz,4H),2.23(s,3H)。
1.2.7 成马来酸盐
将马来酸(2.38 g,0.02 mol)用95%乙醇(10 mL)溶清待用。将95%乙醇(60 mL)和Avitinib(10 g,0.02 mol)投入100 mL 带有机械搅拌的四口瓶中,搅拌升温溶清,溶清时反应体系温度约为60 ℃,控温55 ℃~60 ℃,于快速搅拌下缓慢将已经配置好的马来酸95%乙醇溶液滴入反应体系,滴入过程可见反应体系逐渐变浑浊,滴毕,于55 ℃~60 ℃下保温搅拌1 h,保温毕,搅拌降温至10 ℃以下,抽滤,滤饼用适量95%乙醇洗涤,于80 ℃真空干燥,得到淡黄色细小颗粒状固体11.09 g,为艾维替尼马来酸盐,摩尔收率为89.58%。
2 结果与讨论
2.1 2,4-二氯-7H-吡咯并[2,3-d]嘧啶上保护
因专利US 20150210702 中用2-(三甲基硅烷基)乙氧甲基氯上保护收率只有53.4%,脱保护也较困难且收率不高,故本文直接淘汰该保护方法。
本文除了上乙酰基保护外,还尝试起始原料1-a 在丙酮中用碳酸钾与氯苄反应上苄基保护,收率接近定量转化,且在乙醇中用Pd/C 加氢脱保护,收率也接近定量转化,同时加氢过程硝基也被还原成氨基,转化率很高,但是考虑到加氢工艺被安监总管三[2009]116 号文件定为“高危工艺”,且化工企业氢气储存量超过5 t 就是安全意义上的“重大危险源”,故本文出于工业化的考虑,不建议起始原料1-a 上苄基保护。
本文也尝试1-a 不上保护进行后续反应,发现副反应太多,故不上保护的思路不可取。此处需要强调的是,以起始原料1-a 本身做缚酸剂,反应完毕以其盐酸盐形式分离出来,下一批投料时只需液碱中和后,用二甲苯萃取、干燥后即可继续与乙酰氯反应。
2.2 改用间硝基苯酚钠替代间硝基苯酚
本文以1-b 与间硝基苯酚在二甲苯溶剂中用碱进行反应,尝试了碳酸钾、氢氧化锂、氢氧化钠、叔丁醇钠、三乙胺作为碱,反应效果都不理想。重复专利US 20150210702 中以DMF 为溶剂,碳酸钾为碱的反应方式,液相监控时发现副反应较多,平均收率只有70%左右。后来出于尽量减少溶剂种类的考虑,尝试以二甲苯和水在相转移催化剂的作用下进行两相反应,实验效果竟然出乎所料的好。此外,因该步反应仍以二甲苯为溶剂,尝试将前一步的产物1-b 不分离直接进行反应,发现收率并不受影响。
2.3 Buchwald-Hartwig 交叉偶联反应改进
专利US 20150210702 中关于该步反应的收率并未报道,本文重复后发现收率虽然超过90%,但需引入新溶剂叔丁醇,用二甲苯替代叔丁醇发现收率不足80%,于是进行催化剂、配体和碱的筛选,最终发现1.2.4 章节中叙述的反应条件最佳,仍用二甲苯为溶剂的突出的好处是2-a 也不必分离出来。
2.4 硝基还原方法的选择
本文尝试用专利US 20150210702 中乙醇、还原铁粉、氯化铵的反应条件,发现硝基还原效果较好,但仍需将氨基物分离出来寻找新的方法脱乙酰基保护,步骤较为繁琐。于是采用冰乙酸、还原铁粉的方法先进行硝基还原,产生的副产物铁渣尝试用于污水站芬顿氧化工段与硫酸亚铁混合使用,小试发现降低废水COD 效果较好。滤除铁渣后的氨基物冰乙酸溶液只需加入适量的浓盐酸即可脱乙酰基保护且效果较好,值得一提的是乙酰基脱保护后也是冰乙酸,故3-b 的滤液考虑制备乙酸钠是可行的。
2.5 艾维替尼成马来酸盐过程
用无水乙醇和95%乙醇分别做溶剂进行Avitinib 与马来酸的成盐试验,发现均可得到淡黄色细小颗粒状固体,区别是无水乙醇制得的艾维替尼马来酸盐熔点为201.5 ℃~203.9 ℃,95%乙醇制得的艾维替尼马来酸盐熔点为180.5 ℃~182.7 ℃,另外用智能自动水分滴定仪发现95%乙醇制得的艾维替尼马来酸盐含有5.8%~7.6%的水分,推测95%乙醇制得的艾维替尼马来酸盐分子结构上可能带有两个结晶水,与文献[16]吻合。
3 结论
本文所述的艾维替尼合成工艺较文献更为简便,上保护、醚化两个工序所得产物不需分离就可以直接进入下一步偶联工序。除硝基还原、成马来酸盐工序外,其余反应都采用价廉易得、回收率高的二甲苯为溶剂,溶剂单一在工业上占有优势。此外,乙酰氯替代2-(三甲基硅烷基)乙氧甲基氯使生产成本大幅减少,操作上弹性空间也更大。值得一提的是,本文将硝基铁粉还原的副产物铁渣用于废水处理不失为一个大胆的尝试,既解决了废渣难处理的问题,也节省了废水处理用药剂。因此,本文所述的艾维替尼合成工艺安全环保可控、可操作性强、原料成本降低,可为艾维替尼生产厂家提供一定的参考。
