苹果U6启动子的克隆及功能分析
2020-01-14卞书迅韩晓蕾袁高鹏张利义田义张彩霞丛佩华
卞书迅,韩晓蕾,袁高鹏,张利义,田义,张彩霞,丛佩华
苹果U6启动子的克隆及功能分析
卞书迅,韩晓蕾,袁高鹏,张利义,田义,张彩霞,丛佩华
(中国农业科学院果树研究所/农业部园艺作物种质资源利用重点实验室/国家苹果育种中心,辽宁兴城 125100)
【】U6启动子是CRISPR/Cas9基因组编辑载体系统中驱动sgRNA转录的重要元件,其可能存在物种特异性因子,且长度不同转录活性存在差异。迄今在苹果(×)上对U6启动子尚缺乏研究。因此,筛选出转录活性高且片段大小合适的苹果U6启动子,可以优化苹果CRISPR/Cas9基因编辑体系。利用软件DNAMAN以及启动子元件在线分析网站PLACE和plant CARE对苹果U6启动子进行比对分析;克隆并构建U6启动子驱动萤火虫荧光素酶基因(Firefly luciferase,)的融合表达载体,利用农杆菌介导的瞬时转化法分别转染苹果愈伤组织和本氏烟草()叶片;通过检测荧光素酶活性对各U6启动子进行转录活性比较。苹果基因组中共检索到6条U6 snRNA(E-value<3e-40),分别位于第6、7、9、10、15和17号染色体上,取5′端27 bp snRNA及其上游1 500 bp作为候选U6启动子。序列比对结果显示,苹果U6启动子与拟南芥相同,均具有两个保守的元件,包括上游序列元件(Upstream sequence element,USE)和TATA-Like box。瞬时转化后荧光素酶活性检测结果显示,10号染色体上的U6启动子转录活性最高,10号染色体上5′端截短的U6启动子(长度分别为1 500、959、275和116 bp)中275 bp的启动子活性最强。另外,在苹果愈伤组织中,苹果U6启动子的转录活性要显著高于拟南芥U6启动子。从苹果基因组克隆6条U6启动子,并筛选出一条转录活性高且片段长度较短的U6启动子。
苹果;U6启动子;荧光素酶;本氏烟草;愈伤组织
0 引言
【研究意义】人工优化后的II型CRISPR/Cas9基因编辑体系[1-2],因操作简单、效率高,以及脱靶效应低等优点受到全世界的广泛关注。目前,该系统已应用于多种植物[3-4],在植物分子育种中展现出巨大的前景。具有双链断裂能力的Cas9蛋白和一个人工融合的小向导RNA(single guide RNA)构成了常规的CRISPR/Cas9基因编辑体系[5],其中sgRNA起到靶向结合目的位点的功能,因此,筛选合适的启动子来驱动sgRNA的转录对CRISPR/Cas9基因编辑体系发挥功能有至关重要的意义。【前人研究进展】U6 snRNA(Small nuclear RNA)是一种小的非编码RNA,全长102 bp且序列非常保守[6],受RNA聚合酶III的识别和转录,广泛参与细胞核中mRNA前体(pre-mRNA)的剪接成熟[7-9]。驱动U6 snRNA转录的U6启动子常作为CRISPR/Cas9基因编辑载体中驱动sgRNA转录的重要元件。U6启动子在真核生物中发挥作用需具备两个重要的序列元件USE(Upstream sequence element)和TATA-Like box[6,10],均处于转录起始位点之前[11-12]。此外,U6启动子具有明确的转录起始位点‘G’碱基,可以精确转录sgRNA[13],但精确的起始转录要求转录起始位点周围序列保持稳定[14],前人[15-16]对携带5′端不同长度snRNA(+1 bp,+19—+27 bp)的U6启动子进行了转录活性比较,发现携带27 bp snRNA序列的U6启动子转录活性较高。目前,已有多种植物的U6启动子应用于CRISPR/Cas9基因编辑体系[17-21],LI等[3]和FENG等[22]利用拟南芥中已克隆的启动子驱动sgRNA的转录,在拟南芥中成功建立CRISPR/Cas9基因组编辑体系,并成功敲除拟南芥中和。JIANG等[17]克隆水稻U6启动子驱动sgRNA的转录,并在水稻原生质体中敲除和。JACOBS等[23]克隆了大豆U6启动子,构建靶向敲除绿色荧光蛋白(GFP)基因的Cas9/sgRNA载体,并成功修饰9个大豆内源基因。NISHTANI等[24]利用拟南芥U6启动子驱动sgRNA的转录,成功突变了苹果叶绿素合成相关基因。【本研究切入点】虽然多种植物的U6启动子在CRISPR/Cas9基因编辑体系中得到应用,但U6启动子在亲缘关系较远的物种中并不适用[6],LI等[18]发现仅拟南芥U6启动子具备保守的序列元件‘CAT框’,推测其可能是拟南芥U6启动子的物种特异性因子。并且,不同序列长度、不同组织类型对U6启动子的转录活性也存在一定影响[18,25-26]。目前,还未有苹果内源U6启动子介导的CRISPR/Cas9基因编辑体系。【拟解决的关键问题】对苹果U6启动子的转录活性比较,筛选到长度合适且转录活性高的苹果U6启动子,以期为进一步发展苹果CRISPR/ Cas9基因编辑技术奠定基础。
1 材料与方法
试验于2018—2019年在中国农业科学院果树研究所/农业部园艺作物种质资源利用重点实验室/苹果育种中心实验室进行。
1.1 试验材料
试验所用‘金冠’苹果幼嫩叶片和本氏烟草种子于中国农业科学院果树研究所种质资源圃及实验室中保存;质粒由新西兰皇家植物和食品研究所博士惠赠;(Accession No. KR029101.1)质粒由华南农业大学刘耀光院士惠赠;pTOPO-Blunt Clone kit购于北京艾德莱生物科技有限公司;Prime STAR MAX高保真DNA聚合酶购于TaKaRa公司;DH5α和农杆菌GV3101感受态购于庄盟生物科技有限公司;荧光素钠盐购于上海生工生物工程公司;测序及引物合成由天津金唯智生物公司完成。
1.2 苹果U6启动子克隆及表达载体构建
用拟南芥保守的102 bp U6 snRNA序列在蔷薇科植物基因组数据库GDR(http://www.rosaceae.org/ node/1)中检索苹果候选的U6启动子,在基因组共检索到超过10条U6 snRNA,选择E-value<3e-40的6条,分别取其5′端27 bp U6 snRNA以及上游1 500 bp作为候选苹果U6启动子全长序列,设计引物进行扩增(表1)。用植物基因组DNA提取试剂盒对苹果品种‘金冠’基因组DNA进行提取,使用Prime STAR MAX高保真DNA聚合酶对苹果U6启动子进行扩增。电泳检测正确的片段与pTOPO-Blunt载体连接,转化DH5α感受态测序后进行质粒提取。将T-MdU6-Ps和质粒进行双酶切鉴定(酶切位点见表1),确定正确后用T4 DNA连接酶16℃连接过夜,转化DH5α感受态并提取质粒。为比较苹果和拟南芥U6启动子的转录活性,本试验以质粒为模板克隆长度为301 bp的启动子(表1),并连接至载体,命名为::。不同U6启动子驱动荧光素酶基因的融合表达载体示意图如图1。

图1 U6启动子驱动LUC报告基因的融合表达载体构建

表1 本研究使用的引物序列
小写字母为限制性酶切位点序列 Restriction site sequences are indicated by lowercase letters
1.3 MdU6启动子序列元件分析
利用DNAMAN软件对BLAST检索到的6条苹果U6启动子进行序列比对,分析其保守的序列元件。利用PLACE和Plant CARE启动子在线分析网站预测苹果U6启动子序列元件。
1.4 本氏烟草种植
本氏烟草种子于湿润环境中避光发芽后移入土中(蛭石﹕腐殖土﹕田园土=1.5﹕1﹕1)。温度23℃,光照强度12 000 lx,湿度70%,16 h光照/8 h黑暗条件下培养4—6周,选取第4片之后的功能叶进行注射。
1.5 本氏烟草叶片中瞬时表达分析
将鉴定正确的表达载体转化至农杆菌GV3101感受态。于液体YEB培养基中摇至OD600值为0.8—1.0,离心收集菌体,重悬浮于注射缓冲液(10 mmol∙L-1MgCl2+200 μmol∙L-1AS)中。将重悬液于28℃静置3 h,注射本氏烟草叶片,避光过夜后于23℃光照培养箱进行16 h光照/8 h黑暗培养96 h。每片叶为一次重复,3次重复。
1.6 苹果愈伤组织中瞬时表达分析
方法同1.5,待目的菌液摇至OD600值为0.6—0.8,离心收集菌体,重悬浮于注射缓冲液中,28℃静置活化3 h,取状态良好的苹果‘王林’愈伤组织放入其中,于28℃震荡侵染15 min,无菌水冲洗后用滤纸吸干,置于愈伤培养基中28℃避光共培养96 h,再次冲洗并吸水后放于无抗性愈伤培养基上,重复3次。
1.7 生物发光检测
对瞬时转化96 h后的植物组织的荧光信号进行检测。将植物材料与100 mmol∙L-1荧光素钠盐进行反应,烟草叶片浸泡3—5 min,苹果愈伤组织浸泡10 min。通过高分辨力制冷CCD检测系统进行数据采集(Tanon 5200Multi,中国),运用成像软件对采集的数据进行分析,将数据转换成彩色图像。
1.8 数据分析
试验数据采用Excel 2016进行分析, 并采用SPSS 18.0进行单因素方差分析(<0.05)。
2 结果
2.1 MdU6启动子序列比对分析
基于‘金冠’苹果基因组中的BLAST结果,对E-value<3e-40的6条U6 snRNA进行分析,发现其分别位于苹果第6、7、9、10、15和17号染色体上(表2),全长均为102 bp且序列保守,另外,其转录起始位点均为‘G’碱基。参照PAUL等[15]的结论,本试验选取各U6 snRNA的5′端27 bp及其上游 1 500 bp作为苹果U6启动子候选序列,分别命名为MdU6-6P、MdU6-7P、MdU6-9P、MdU6-10P、MdU6- 15P和MdU6-17P。选取转录起始位点前后150 bp序列进行比对,发现其均存在-30位点的TATA-like box和-60位点的USE(5′-GTCCCACATCG-3′),且两个元件之间的距离较为保守(图2),其中TATA- like box与RNA聚合酶III的识别和结合相关。
表2 苹果U6 snRNA染色体定位

图2 MdU6启动子序列分析
2.2 苹果U6启动子的克隆及鉴定
从‘金冠’基因组DNA中成功克隆出MdU6-6P、MdU6-7P、MdU6-9P、MdU6-10P、MdU6-15P和MdU6-17P,测序结果显示序列正确。将各启动子片段分别连入载体,并命名为::、::、::、::、::和::(图3)。
2.3 苹果U6启动子转录活性鉴定
将各::载体通过农杆菌介导转化法注射于烟草叶片,并以::为阳性对照,空载体为阴性对照。96 h后进行荧光素酶活性检测,发现MdU6-10P荧光信号最强,MdU6-9P仅次于MdU6-10P,而MdU6-6P、MdU6-7P、MdU6-15P以及MdU6-17P的荧光信号均较弱(图4)。
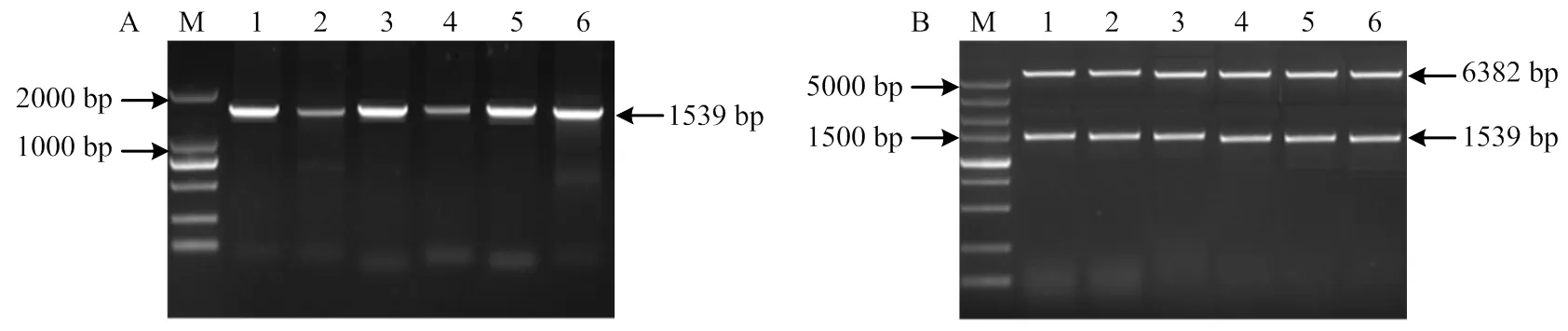
A:M:2 kb DNA标准分子量M: DL2000 DNA marker; 1: MdU6-6P; 2: MdU6-7P; 3: MdU6-9P; 4: MdU6-10P; 5: MdU6-15P; 6: MdU6-17P: B:M:5 kb DNA标准分子量M: DL5000 DNA marker; 1: MdU6-6P::LUC; 2: MdU6-7P::LUC; 3: MdU6-9P::LUC; 4: MdU6-10P::LUC; 5: MdU6-15P::LUC; 6: MdU6-17P::LUC
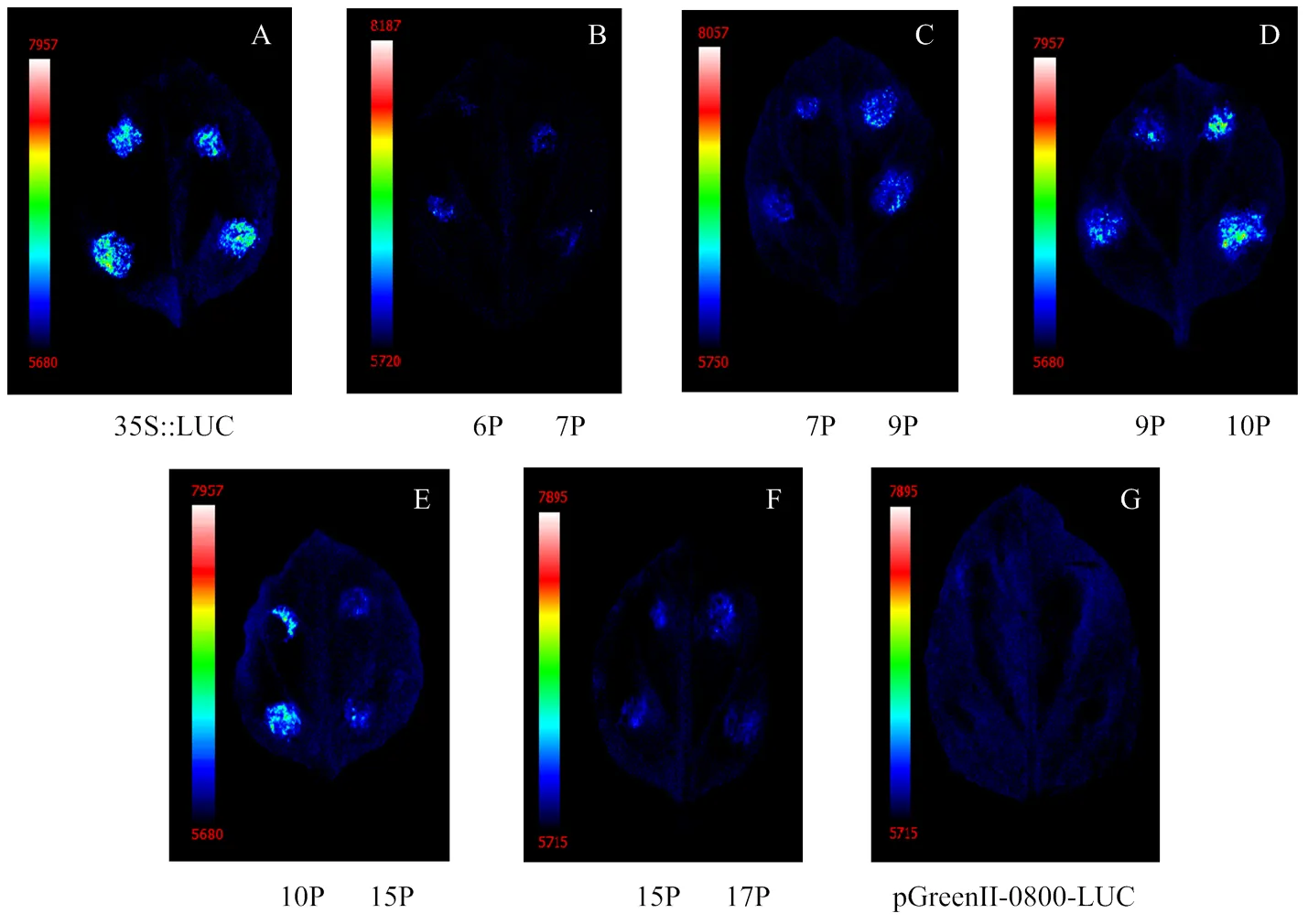
6P: MdU6-6P::LUC; 7P: MdU6-7P::LUC; 9P: MdU6-9P::LUC; 10P: MdU6-10P::LUC; 15P: MdU6-15P::LUC; 17P: MdU6-17P::LUC
为比较MdU6-9P和MdU6-10P在苹果中的转录活性,进一步将::和::载体瞬时转化苹果愈伤组织,对荧光素酶活性进行检测并将荧光信号数据进行统计分析(图5)。发现::的荧光信号强度显著高于::,证明MdU6-10P具有较高的转录活性,因此,选取苹果10号染色体上的U6启动子用于后续试验。

不同小写字母表示差异显著(P<0.05)。下同 Different lowercase letters indicate significant difference (P<0.05). The same as below
2.4 MdU6-10P元件分析
为分析序列长度对MdU6-10P转录活性的影响,使用PLACE和Plant CARE启动子元件在线分析网站对MdU6-10P进行元件分析,以预测的‘CAT框’为依据,对其进行5′端截短(截短长度分别为959、275和116 bp)。截短后的苹果U6启动子分别命名为MdU6-10-2P、MdU6-10-3P和MdU6-10-4P(图6)。
2.5 MdU6-10-Ps的克隆及鉴定
从‘金冠’苹果基因组中成功克隆出MdU6-10-2P、MdU6-10-3P和MdU6-10-4P,将各启动子片段分别连至载体,将测序正确的各载体分别命名为::、::C和::(图7)。
2.6 MdU6-10-Ps转录活性鉴定
利用农杆菌介导转化法,将::、、::和-::载体注射烟草叶片。检测荧光素酶活性并进行分析,发现注射后的烟草叶片均具有较强的荧光信号,但并无显著差异(图8)。

图6 5′端截短的苹果U6启动子示意图
进一步在苹果愈伤组织中验证各长度苹果U6启动子转录活性,荧光素酶活性检测结果与烟草叶片不同,如图9所示。MdU6-10-Ps转染后的愈伤组织荧光信号强度存在显著差异(MdU6-10-3P>MdU6-10-2P>MdU6-10P>MdU6-10-4P),其中,MdU6-10-3P荧光信号最强,而MdU6-10-4P荧光信号强度最弱,但仍可检测到荧光信号。
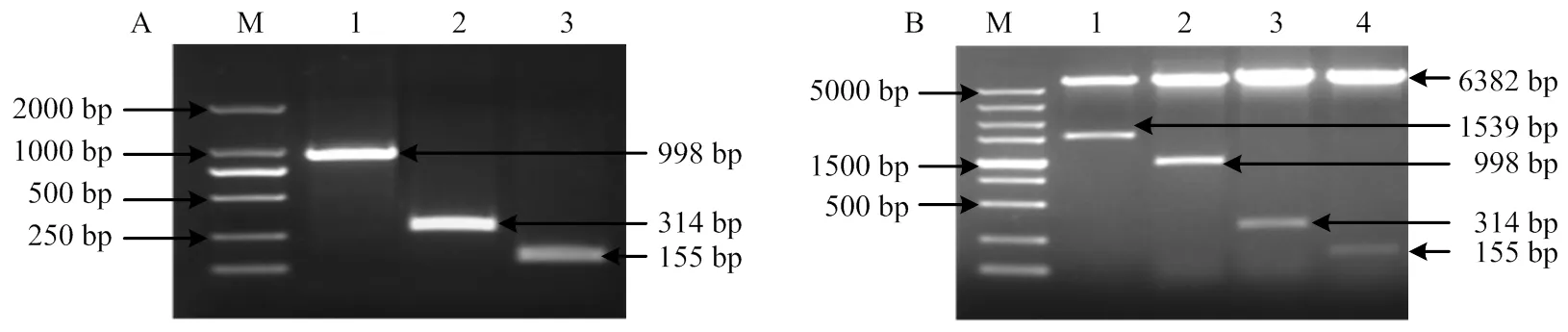
A:M:2 Kb DNA 标准分子量M: DL2000 DNA marker; 1: MdU6-10-2P; 2: MdU6-10-3P; 3: MdU6-10-4P; B:M:5Kb DNA标准分子量M: DL5000 DNA marker; 1: MdU6-10P::LUC; 2: MdU6-10-2P::LUC; 3: MdU6-10-3P::LUC; 4: MdU6-10-4P::LUC

A: MdU6-10P::LUC; B: MdU6-10-2P::LUC; C: MdU6-10-3P::LUC; D: MdU6-10-4P::LUC
2.7 苹果和拟南芥U6启动子转录活性比较
为比较拟南芥和苹果U6启动子在苹果中的转录活性,将::和::载体分别瞬时转化至苹果愈伤组织,结果如图10所示。::转染的愈伤组织荧光信号强度显著高于::。说明在苹果愈伤组织中,苹果U6启动子的转录活性高于拟南芥U6启动子。
3 讨论
RNA介导的第3代人工核酸酶技术CRISPR/Cas9在植物基因功能验证及定向育种中展现出巨大的潜力,但由于序列较长(大于4 000 bp),且驱动转录的35S启动子长度也有800 bp,如果构建同时编辑多个靶点的CRISPR/ Cas9基因编辑载体,序列长度将达到15 000 bp,载体过长将会对细菌转化和植物组织转染造成困难。因此,筛选合适的启动子驱动sgRNA转录将为该体系的应用提供便利。U6启动子常被用来驱动sgRNA的转录。U6启动子属于RNA聚合酶III识别的启动子,III型启动子主要是对tRNA、5S rRNA等含发夹结构的小RNA进行转录[8],因此,U6启动子在转录含发夹结构的sgRNA时具有天然优势,并且可以确保转录的sgRNA在细胞核中发挥功能。

图10 AtU6-1P::LUC和MdU6-10-3P::LUC在苹果愈伤组织中瞬时表达
此外,U6启动子具有精确的转录起始位点‘G’碱基,可以有效降低CRISPR/Cas9基因编辑体系的脱靶效应。本试验将拟南芥启动子和苹果6条候选U6启动子进行序列比对,发现6条候选的苹果U6启动子的转录起始位点均为‘G’碱基,且均具有RNA聚合酶III发挥作用所需的上游序列元件USE和TATA-like box。由于U6 snRNA序列高度保守,通过荧光定量PCR来比较各U6启动子的转录活性并不可行,因此,本试验通过利用U6启动子驱动报告基因表达的方式对苹果U6启动子的转录活性进行比较。目前,常用于启动子活性检测的报告基因有[27][28-29]和[30],本试验选择为报告基因,通过瞬时转化后检测荧光素酶活性对各启动子的转录活性进行比较,发现所选的6条苹果U6启动子均具有转录活性,但转录活性存在差异,其中MdU6-10P具有较高的转录活性。前人研究表明,U6启动子在具备必要元件时即具有转录活性,反而序列过长可能存在一些抑制因子,使较长的U6启动子的转录活性下降[18]。在番茄[21]上,U6启动子被截取到200 bp左右时仍具有较高的活性,而在棉花[31]上,U6启动子截取到105 bp仍具有较高的活性。KHAOULA等[32]将拟南芥U6启动子截取到79 bp仍可应用于基因编辑体系。本研究对苹果10号染色体上的U6启动子进行5′端截短,并进行转录活性比较,发现在苹果愈伤组织中,长度为275 bp的苹果U6启动子转录活性最高,表明在苹果U6启动子中可能存在一些抑制因子,且存在于5′末端。荧光素酶活性检测结果还显示,116 bp长度的U6启动子活性虽减弱,但并未丧失转录活性,说明苹果U6启动子具有必要元件即有转录活性,而在序列较短时转录活性下降可能是因为缺少了一些增强转录活性的元件。至于在本氏烟草和苹果愈伤组织中瞬时转染展现出不同的结果,或许是因为某些物种特异性因子参与其中,导致U6启动子在亲缘关系较远的物种中转录活性存在一定差异,这与前人得出的结论一致[6]。植物的不同组织类型可能对U6启动子的转录活性也存在一定影响[18],而苹果不同组织类型对U6启动子转录活性的影响程度还有待进一步研究。
4 结论
从苹果品种‘金冠’中克隆获得6条长度为1 500 bp的U6启动子,均具有转录活性,其中10号染色体上的U6启动子转录活性最高,将该启动子从5′端进行截短(长度分别为959、275和116 bp),其中275 bp的启动子转录活性最高。此外,在苹果愈伤组织中,苹果U6启动子的转录活性高于拟南芥U6启动子。
[1] CHEN K L, GAO C X. Targeted genome modification technologies and their applications in crop improvements., 2014,33(4): 575-583.
[2] RAN F A, HSU P D, WRIGHT J, AGARWALA V, SCOTT D A, ZHANG F. Genome engineering using the CRISPR-Cas9 system., 2013,8(11): 2281-2308.
[3] LI J F, NORVILLE J E, AACH J, MCCORMACK M, ZHANG D D, BUSH J, CHURCH G M, SHEEN J. Multiplex and homologous recombination-mediated genome editing inandusing guide RNA and Cas9., 2013,31(8): 688-691.
[4] LIU X J, XIE C X, SI H J, YANG J X. CRISPR/Cas9-mediated genome editing in plants., 2017,12(1): 94-102.
[5] XING H L, DONG L, WANG Z P, ZHANG H Y, HAN C Y, LIU B, WANG X C, CHEN Q J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants., 2014,14(1): 327.
[6] DAS G, HENNING D, REDD R. Structure, organization, and transcription of Drosophila U6 small nuclear RNA genes., 1987,262(25): 1187-1193.
[7] TAZI J, FORNE T, JEANTEUR P, CATHALA G, BRUNEL C. Mammalian U6 small nuclear RNA undergoes 3' end modifications within the spliceosome., 1993,13(3): 1641-1650.
[8] BERGET S M, ROBBERSON B L. U1, U2, and U4/U6 small nuclear ribonucleoproteins are required for in vitro splicing but not polyadenylation., 1986,46(5): 691-696.
[9] BLACK D L, STEITZ J A. Pre-mRNA splicing in vitro requires intact U4/U6 small nuclear ribonucleoprotein., 1986,46(5): 697-704.
[10] WAIBEL F, FILIPOWICZ W. U6 snRNA genes ofare transcribed by RNA polymerase III but contain the same two upstream promoter elements as RNA polymerase ll-transcribed U-snRNA genes., 1990,18(12): 3451-3458.
[11] BOGENHAGEN D F, SAKONJU S, BROWN D D. A control region in the center of the 5S RNA gene directs specific initiation of transcription: II. The 3' border of the region., 1980,19(1): 27-35.
[12] GEIDUSCHEK E P, TOCCHINI-VALENTINI G P. Transcription by RNA polymerase III., 1988,57(59): 873-914.
[13] MALI P, YANG L H, ESVELT K M, AACH J, GUELL M, DICARLO J E, NORVILLE J E, CHURCH J M. RNA-guided human genome engineering via Cas9., 2013,339(6121): 823-826.
[14] MA H M, WU Y G, DANG Y, CHOI J G, ZHANG J L, WU H Q. Pol III promoters to express small RNAs: Delineation of transcription initiation., 2014,3(5): e161.
[15] PAUL C P, GOOD P D, WINER I, ENGELKE D R. Effective expression of small interfering RNA in human cells., 2002,20(5): 505-508.
[16] KWAK Y D, KOIKE H, SUGAYA K. RNA interference with small hairpin RNAs transcribed from a human U6 promoter-driven DNA vector., 2003,93(2): 214-217.
[17] JIANG W Z, ZHOU H B, BI H H, FROMM M, YANG B, WEEKS D P. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in, tobacco, sorghum and rice., 2013,41(20): e188.
[18] LI X, JIANG D H, YONG K, ZHANG D B. Varied transcriptional efficiencies of multiplesmall nuclear RNA genes., 2007,49(2): 222-229.
[19] SHAN Q W, WANG Y P, LI J, ZHANG Y, CHEN K L, LIANG Z, ZHANG K, LIU J X. Targeted genome modification of crop plants using a CRISPR-Cas system., 2013,31(8): 686-688.
[20] 李继洋, 雷建峰, 代培红, 姚瑞, 曲延英, 陈全家, 李月, 刘晓东. 基于棉花 U6 启动子的海岛棉CRISPR/Cas9基因组编辑体系的建立. 作物学报, 2018,44(2): 227-235.
LI J Y, Lei J F, Dai P H, Yao R, Qu Y Y, Chen Q J, Li Y, Liu X D. Establishment of CRISPR/Cas9 genome editing system based on GbU6 promoters in cotton (L.)., 2018, 44(2): 227-235. (in Chinese)
[21] 蒲艳, 刘超, 李继洋, 阿尔祖古丽·塔什, 胡燕, 刘晓东. 番茄 U6 启动子的克隆及 CRISPR/Cas9 基因编辑体系的建立. 中国农业科学, 2018, 51(2): 315-326.
PU Y, Liu C, Li J Y, Alzu G T, Hu Y, Liu X D. Differentpromoters cloning and establishment of CRISPR/Cas9 mediated gene editing system in tomato., 2018, 51(2): 315-326. (in Chinese)
[22] FENG Z Y, ZHANG B T, DING W N, LIU X D, YANG D L, WEI P L. Efficient genome editing in plants using a CRISPR/Cas system., 2013,23(10): 1229-1232.
[23] JACOBS T B, LAFAYETTE P R, SCHMITZ R J, PARROTT W A. Targeted genome modifications in soybean with CRISPR/Cas9., 2015,15(1): 1-10.
[24] NISHITANI C, HIRAI N, KOMORI S, WADA M, OKADA K, OSAKABE K, YAMAMOTO T, OSAKABE Y. Efficient genome editing in appleusing a CRISPR/Cas9 system., 2016,6: 31481.
[25] 朱金洁. CRISPR_Cas9介导的玉米基因组定点编辑研究[D]. 北京:中国农业大学, 2015.
ZHU J J. Targeted genome editing in maize using CRISPR-Cas9 [D]. Beijing: China Agricultural University, 2015. (in Chinese)
[26] WANG M B, HELLIWELL C A, WU L M, WATERHOUSE P M, PEACOCK W J, DENNIS E S. Hairpin RNAs derived from RNA polymerase II and polymerase III promoter-directed transgenes are processed differently in plants., 2008,14(5): 903-913.
[27] JEFFERSON R A, KAVANAGH T A, BEVAN M W. GUS fusions: Beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants., 1987,6(13): 3901-3907.
[28] CHIU W L, NIWA Y, ZENG W K, HIRANO T, KOBAYASHI H, SHEEN J. Engineered GFP as a vital reporter in plants., 1996,6(3): 325-330.
[29] ZHANG G H, GURTU V, KAIN S R. An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells., 1996,227(3): 707-711.
[30] WELSH S, KAY S A. Reporter gene expression for monitoring gene transfer., 1997,8(5): 617-622.
[31] 雷建峰. 棉花U6启动子克隆与功能分析及拟南芥GGB突变体的创制[D]. 乌鲁木齐:新疆农业大学, 2016.
LEI J F. Cloning and functional analysis of U6 promoters in cotton and creation ofGGB mutant [D]. Urumqi: Xinjiang Agricultural University, 2016. (in Chinese)
[32] KHAOULA B, ANGELA C G, SOPHIEN K, NEKRASOV V. Plant genome editing made easy: Targeted mutagenesis in model and crop plants using the CRISPR/Cas system., 2013,9(1): 39.
Cloning and Functional Analysis of U6 Promoter in Apple
BIAN ShuXun, HAN XiaoLei, YUAN GaoPeng, ZHANG LiYi, TIAN Yi, ZHANG CaiXia, CONG PeiHua
(Institute of Pomology, Chinese Academy of Agricultural Sciences/Key Laboratory of Fruit Germplasm Resources Utilization, Ministry of Agriculture/National Apple Breeding Center, Xingcheng 125100, Liaoning)
【】U6 promoter is an important element for the transcription of sgRNA in the CRISPR/Cas9 genome editing system. There may be species-specific factors in U6 promoter, and the activity of U6 promoter would be altered when its length changed. So far, in apple (), the transcriptional characterization of U6 promoters has not been reported. Therefore, selecting an apple U6 promoter with high transcriptional activity and suitable fragment size would provide a basis for optimizing apple CRISPR/Cas9 gene editing technology system. 【】DNAMAN, promoter cis element online predicting website PLACE and plant CARE were used to do the comparative analysis of apple U6 promoters; U6 promoters were cloned and constructed into firefly luciferase vector; the apple callus and tobacco () leaves were transformed via-mediated transient transformation; the transcriptional activity of U6 promoter was determined according to the luciferase activity. 【】There were six alternative U6 promoters in apple genome (E-value<3e-40), which were located on chr 6, chr 7, chr 9, chr 10, chr 15 and chr 17, respectively. 27 bp snRNA at 5′ end and its upstream 1 500 bp were selected as candidate U6 promoter. Sequence analysis results showed that the upstream sequence element (USE) and TATA-like elements were contained in six U6 promoters of apple, the same as. After transient transformation, the luciferase activity assay showed that, the U6 promoter on chromosome 10 had the highest transcriptional activity. Among all U6 promoters which were shortened at 5′ end (1 500 bp, 959 bp, 275 bp, and 116 bp) on chromosome 10, the 275 bp one had the highest transcriptional activity. In addition, compared with the Arabidopsis U6 promoter, in apple callus, the transcriptional activity of the apple U6 promoter was higher. 【】Six U6 promoters were cloned from the apple genome, and a U6 promoter with high transcriptional activity and suitable sequence fragment was obtained.
; U6 promoter; luciferase;; callus

10.3864/j.issn.0578-1752.2019.23.016
2019-07-31;
2019-09-19
国家现代农业产业技术体系建设专项资金(CARS-27)、中国农业科学院科技创新工程(CAAS-ASTIP-2016-RIP -02)、辽宁省博士科研启动基金(20180540030)、中央级公益性科研院所基本科研业务费专项(1610182019007)
卞书迅,E-mail:bb18236767941@163.com。韩晓蕾,E-mail:hanxiaolei@caas.cn。卞书迅与韩晓蕾为同等贡献作者。
张彩霞,Tel:0429-3598135;E-mail:cxzhang-bj@163.com。通信作者丛佩华,Tel:0429-3598103;E-mail:congph@163.com
(责任编辑 赵伶俐)
