First-principles Calculations on Electrical Properties Effect of Li on Graphene, BC5, C5N Surface
2020-01-13
(School of Mining and Coal, Inner Mongolia University of Science & Technology, Baotou 014010, China)
Abstract:The geometrical structures of Li atom adsorption on the most stable sites of pristine graphene and BC5, C5N surfaces were optimized by using first-principles method based on density functional theory (DFT). The band structures, density of states (DOS), charge transferring, electron density differences and binding energies of both pristine graphene and BC5, C5N were calculated theoretically. The results revealed that when the B-doping concentration is 16.67%, the adsorbing energy of Li atom on graphene will be remarkably enhanced; however, when the N-doping concentration is 16.67%, the adsorbing energy of Li atom on graphene will be decreased. GraphenE-Li, BC5-Li and C5N-Li all have metal properties. There is mixed ionic and covalent bonds between Li and graphene, BC5 and C5N.
Key words: first-principle; graphene; boron (Nitrogen)-doped graphene; adsorption of Li atom; electronic structure
1 Introduction
Lithium-ion batteries are now widely used in portable electronic devices and are potential power for electric vehicles. Lithium intercalation compounds and graphite are often used as anode and cathode materials for lithium-ion batteries[1-2]. However, due to the limited lithium storage capacity of graphite (372 mAh/g)[3], it does not meet the requirement for higher lithium storage capacity. Graphene has caused great attention because of its excellent structure and electronic properties[4-5]. Recently, Yoo et al[6]found that the multi-layer graphene nanosheets have a lithium storage capacity of 540 mAh/g, which is 1.5 times than that of graphite. Recent studies have shown that since B and N atoms have adjacent atomic radius with C atom, graphene can be doped by substitution, and boron-doping, nitrogen-doping can effectively improve the lithium storage performance of graphene, which is confirmed theoretically and experimentally. Kuzubov et al[7]used first-principle method found that lithium atom enter the graphitE-like BC3crystals by intercalation to form Li2BC3compounds, and the lithium storage capacity was three times than that of graphite. Zhao Yinchang et al[8]studied the adsorption behavior of lithium atom on both sides of singlE-layer BC7sheet, the calculation results showed that the stoichiometric ratio of lithium atom on both sides of BC7sheet can reach BC7Li7, and the lithium storage capacity was 1977 mAh/g, which is 5.3 times than that of graphite, and the systems before and after lithium adsorption were both good conductor. Li et al[9]found that nitrogen doping improved the specific capacity and cycle performance of graphene nanosheets. The first specific capacity of graphene after nitrogen doping was 684 mAh/g, and the specific capacity after 100 cycles was 452 mAh /g, the lithium storage capacity was still higher than graphite. Zhou et al[10]used density functional theory to study the adsorption of lithium atom on C, BC3, BC2N, BN singlE-walled carbon nanotubes, the adsorption energies of lithium atom on different carbon nanotubes were arranged in the following order:BC3>C>BN>BC2N. It can be seen that the electronic structure had an important effect of lithium adsorption on different nanotubes.
Many scholars have carried out research on the theoretical characteristics of lithium embedded on graphite (graphene) materials from the charge transfer, charge density distribution and band structure[11-12]. Doping graphene is also one of the researching hotspots. Gao Ruiling et al[13]studied the electronic structure of graphene doped with diboron, dinitrogen and boron nitrogen by density functional theory. The results showed that boron and nitrogen doping graphene exhibit p-type and n-type semiconductor properties respectively. Ottaviana et al[14]used first-principles method to predict the electronic properties of BC5compounds. The results showed that the BC5monolayer has metal conductivity.
The electrical property of graphene after boron and nitrogen doping changed, and the lithium storage performance was also different. The BC5material has been proved to have a high lithium storage capacity, and the lithium storage performance of the C5N material remains to be studied. Therefore, in order to better understand the changes of the electronic properties of graphene and BC5, C5N before and after lithium adsorption, we use first-principles method to explain the charge transfer of lithium on the stable adsorption position of intrinsic graphene and BC5, C5N. The effect of lithium adsorption on graphene, BC5and C5N’s charge density distribution was analyzed by calculating the charge density difference before and after lithium adsorption. The electronic band structure and density of states before and after lithium adsorption were calculated to analyze the conductivity effect of lithium adsorption on graphene, BC5and C5N.
2 Calculation model and method
In this paper, we use first-principle method VASP software package[15-16]to calculate, which is based on the density functional theory (DFT) first-principles pseudopotential plane wave method. We use the projector augmented-wave method (PAW)[17]to describe the interaction between ions and valence electrons, and the exchangE-correlation potential is treated by local density approximation (LDA)[18]. The optimized results of plane wave truncation energy is 475 eV. The k grid point of the Monkhorst-Pack[18]type is set to 9×9×1. Take theXandYdirections in the graphene plane, and theZdirection is perpendicular to the graphene plane. To eliminate the interlayer atomic interaction force and avoid the interference of periodic arrangement, the vacuum layer height is 15 Å. Structural relaxation is performed first. The maximum convergence accuracy of each atom in the relaxation process is 0.01 eV/Å, and the total energy error is no more than 1.0×10-4eV. Using Blöchl corrected tetrahedron method, the energy expansion width is 0.05 eV.

Fig.1 The configuration of Li adsorption on pristine and Boron(Nitrogen)-doped graphene
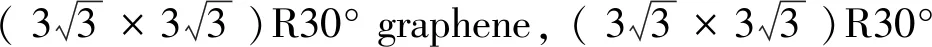
In order to compare the adsorption stability of Li atom on the surface of graphene, BC5and C5N, we calculated the adsorption energy of Li atom at the most stable position on the surface of different graphene systems, and the adsorption energies were obtained by the following formula:
Ead=ELi+Egraphene-Econfig
(1)

In order to reflect the bond formation, charge distribution and charge transfer between atoms, we calculated the differential charge density of Li on different graphene systems, and the differential charge density was obtained by the following formula:
Δρ=ρgraphene-(ρB/N+ρC)
(2)
Δρ=ρLi+graphene-(ρLi+ρgraphene)
(3)
In formula (2)-(3),Δρwas the differential charge density,ρgraphenewas the charge density of BC5/C5N,ρB/Nwas the charge density of boron/nitrogen atom,ρCwas the charge density of carbon atom,ρLi+graphenewas the charge density of Li adsorption on BC5/C5N surface,ρLiwas the charge density of Li atom.
3 Results and discussion
3.1 Differential charge density analysis of Li adsorption on graphene, BC5 and C5N surface
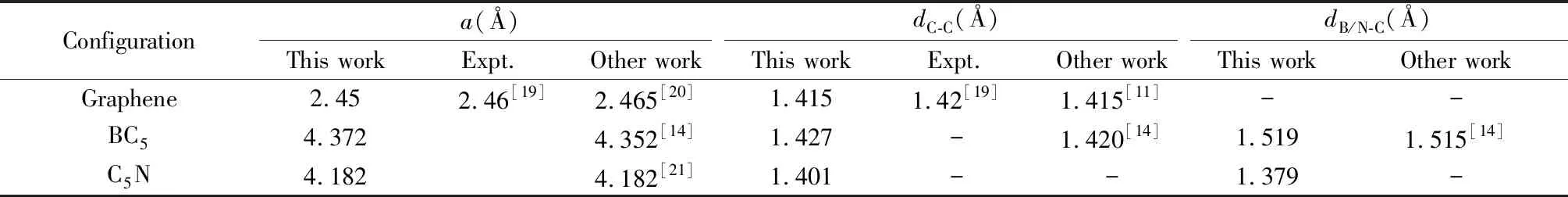
Table 1 The calculation results and relevant experimental values of lattice constant, and the distance between adjacent atoms
The lattice constants of graphene, BC5, and C5N in Table 1 and the distance between adjacent carbon atoms and adjacent boron (nitrogen) atoms with carbon atoms are coincide with experimental values or other scholars’ research results.

Fig.2 The differential charge density of doping graphene
Differential charge densities of B and N doped graphene are shown in
Fig.2 (a) and (b). In contour dense areas get electrons, and in contour sparse areas lose electrons. Because the electronegativity of B, C and N atoms increases in turn, the high-density region between B-C bonds tends to C atoms, forming B-C covalent bond with an atomic spacing of 1.519 Å; the high-density region between N-C bonds tends to N atom, forming N-C covalent bond with an atomic spacing of 1.379 Å. The Bader charge analysis shows that B atom loses electrons, and the charge is transferred from B atom to the bonding C atom, and the transfer amount is 1.2691 e; the N atom obtains electrons, and the transfer amount is 1.2208 e.

Table 2 Performance statistics of one Li atom adsorption on the most stable sites of graphene,BC5 and C5N surface
In Table 2, Site is the most stable adsorption position of Li atom on different graphene surfaces,d⊥is the vertical adsorption height of Li atom on intrinsic graphene, BC5, C5N surfaces,dLi-CanddLi-B/Nare the bond lengths of Li atom with its nearest carbon and boron/nitrogen atoms, Li-C bonds and Li-B/N bonds are the number of bonding of Li atom with C and B/N atoms, respectively.
It can be seen from Table 2 and
Fig.1 that the most stable adsorption position of Li atom on graphene surface is the center of C6ring, while the most stable adsorption positions on BC5and C5N surfaces are the center of BC5ring and C5N ring, which are biased towards the C atom opposite to B atom and N atom. The Li-C bond lengths at the three stable adsorption sites are close to the covalent bond length of Li atom with C atom (2.100 Å), Li-B bond length is close to the covalent bond length of Li atom with B atom (2.210 Å), and Li-N bond length is close to the covalent bond length of Li atom with N atom (2.030 Å), which indicates that the interactions of Li with graphene, BC5and C5N are chemical adsorption. The adsorption energy of Li atom at the H1-site of BC5is 2.083 eV higher than that at the H-site of intrinsic graphene, while the adsorption energy of Li at the H2-site of C5N is 0.603 eV lower than that at the H-site of intrinsic graphene. This indicates that the adsorping energy of Li is enhanced when the boron doping concentration is 16.67% (atomic fraction), while the adsorption energy of Li is weakened when the nitrogen doping concentration is 16.67% (atomic fraction). In addition, boron doping slightly reduces the vertical distance between Li and graphene sheets, on the contrary, nitrogen doping increases the vertical distance between Li and graphene sheets.

Fig.3 The differential charge density of Li adsorption on the most stable sites of pristine and Boron (Nitrogen)-doped graphene. (a),(c),(e)are the side view and top view of Li adsorption on graphene,BC5, C5N, respectively.(b)Plan I-section of
Fig.3(a);(d)Plan II-section of figure 3(b);(f)Plan III-section of
Fig.3(e)
Fig.3 shows the differential charge density of Li on the stable adsorption sites of intrinsic graphene and doped graphene. The positive value in the scale represents an increase in charge density, and the negative value represents a decrease in charge density, that is, the ability of losing electrons from top to bottom is getting stronger. In
Fig.3(a) and (b), there is an obvious phenomenon of charge transfer between Li atom and the nearest C atom. Li is missing a large amount of electrons, and the electronegativity is increasing; while near C is adding a large amount of electrons, the electronegativity is reducing. The Bader charge analysis shows that the charge contribution of Li atom to the nearest carbon atom is 0.8743 e. This is consistent with the results of Holzwarth[11]. In
Fig.3(c) and (d), the charge contribution of Li atom to the adjacent boron and carbon atoms is 0.8694 e. The Li-B bond length is 2.247 Å, and the Li-C bond length is 2.146 Å. The C atom is more prone to obtain electrons, so a greater density of charge accumulation occurs between Li and C atoms. In
Fig.3(e) and (f), the charge contribution of Li atom to the nearest carbon atom is 0.8699 e, and forms three types bonds with the nearest C atom with a bond length of 2.155 Å, 2.175 Å, and 2.207 Å. Therefore there are charge accumulations of different densities between Li and C atoms. Since the doped compounds have different electronic structures, B atom is one electron less than C atom, so BC5is an electron-deficient system, and there is few difference of electronegativity between B and C, forming a polar covalent bond, which will facilitate the adsorption of electron-donating Li atoms; N atom has one more electron than C atom, so single layer C5N is an electron-rich system, which will weaken the adsorption of Li on the surface of C5N.
3.2 Band structure and density analysis of Li adsorption on graphene, BC5 and C5N surfaces
In order to study the effect of electronic structure of Li adsorption on different graphene systems, the band structure and density of states of different graphene systems before and after Li adsorption were compared. In this paper, the energy of 0 eV is defined as the Fermi level, and the band integral path is selected as Γ-K-M-Γ(Fig.4).






4 Conclusion

(1) When B-doped concentration is 16.67% (atomic fraction) it forms an electron-deficient state, which facilitates the adsorption of Li atom with free electrons, and can significantly increase the adsorption energy of Li on graphene; when N-doped concentration is 16.67% (atomic fraction) it forms an electron-rich state, which weakens the adsorption energy of Li on the graphene surface.
(2) After a single Li atom adsorption on the surfaces of graphene, BC5and C5N, the graphenE-Li, BC5-Li and C5N-Li systems exhibit metallicity.
(3) A large amount of charge transfers occur between Li and different doping graphene systems, causing strong ionic bonds occur between Li and graphene, BC5and C5N. In the graphenE-Li system, the 2p orbit of Li atom and the 2p orbit of C atom appear hybridization. In BC5-Li system the 2p orbit of Li atom and the 2p orbits of C1, C2, C3 and B atoms appear hybridization. In C5N-Li system the 2p orbit of Li atom and the 2p orbits of C1, C2, C3, and N atoms appear hybridization, which leads to the covalent bonding of Li and graphene, BC5, and C5N. Therefore, there is a mix of ionic bonds and covalent bonds between Li and graphene, BC5and C5N.
