通过加权基因共表达网络分析(WGCNA)研究食管腺癌的关键基因
2020-01-07王新帅张晨曦李新阳王子明王钰鲲张卫国
姚 歌,王新帅,张晨曦,李新阳,王子明,王钰鲲,张卫国
食管癌是发生在食管上皮组织的恶性肿瘤,是全世界范围内癌症相关死亡的主要原因之一,占所有恶性肿瘤的2%,死亡率高达90%,发生率和死亡率在恶性肿瘤中分别居第六位和第四位[1]。食管癌主要有两种组织学亚型:鳞状细胞癌和腺癌,都是高度侵袭性肿瘤。最新的流行病学数据表明,全球79%的食管鳞癌发生在东南部和中亚,而46%的食管腺癌发生于北欧、西欧、北美洲和大洋洲[2]。在过去的几年中,发达国家腺癌发病率有所上升,而鳞癌的发病率下降。同时食管癌仍然是世界范围内最难治愈的恶性肿瘤之一,由于诊断时多为晚期,只有不到50%的食管癌患者有手术切除的机会。近年来随着手术、化疗和放疗技术的发展,食管癌的临床治疗有了很大进展,但5 a总生存率仍然很低。分子靶向治疗是在基因分子水平上,针对已经明确的致癌位点来设计相应的治疗药物,药物进入体内会特异地结合致癌位点发生作用,使肿瘤细胞特异性坏死,较少对正常组织影响。随着肿瘤分子靶向治疗在肺癌、结直肠癌等疾病的成功应用,加上靶向药物本身选择性高、抗肿瘤活性强优点,分子靶向治疗已成为人们试图解决食管癌治疗问题的新途径。但目前食管癌可应用的靶点较少,因此寻找新的分子靶点显得尤为重要。加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)是鉴定表型性状的基因模块和关键基因的理想方法,已有研究采用此方法对乳腺癌、肺癌、结直肠癌等恶性肿瘤进行分析,但尚未有应用WGCNA对食管腺癌进行分析的研究。因此,本研究采用WGCNA算法识别与食管腺癌发展高度相关的基因模块,并从中筛选出枢纽基因,为进一步发现新的生物标志及食管腺癌诊断和治疗的潜在靶点提供重要的理论依据。现报道如下。
1 材料与方法
1.1 数据来源及数据预处理基因表达谱数据(GSE92396)及其临床信息来源于美国国家生物信息中心下的GEO数据库,平台编号为GPL6244,共纳入22例样本的基因表达数据用于分析,包含9例正常食管组织样本、12例食管腺癌组织及1例介于两者之间的组织样本基因表达谱数据。在进行WGCNA前,对下载的矩阵信息进行了预处理:利用平台信息将探针名转化为基因名,将对应多个探针的基因表达量作log2处理及标准化处理后作为该基因的最终表达量。最终获得行名为样本名,列名为基因名的矩阵用于后续共表达网络的构建。
1.2 数据筛选在R语言下运行WGCNA包,进行共表达网络的构建,为获取有效的共表达网络,计算各基因在所有样本中表达量的方差,取方差前25%的基因用于共表达网络的构建。对样本进行聚类分析,检测并去除离群异常值。
1.3 构建基因共表达网络通过选择一个合适的加权系数β(软阈值)使构建网络中的基因之间的连接服从无尺度网络分布,并利用基因之间的相关系数构建分层聚类树,聚类树的不同分支代表不同的基因模块,不同颜色代表不同的模块。然后基于基因的加权相关系数,将基因按照表达模式进行分类,将模式相似的基因归为一个模块,从而将基因通过基因表达模式归类成不同的模块来进行下一步分析。最后应用此系数将相关矩阵转换为邻接矩阵,进一步转化为拓扑重叠矩阵(Topological Overlap Matrix,TOM),并随机选取400个基因做TOM热图来证明模块之间的高度独立性以及每个模块中基因表达的相对独立性。
1.4 模块与临床特征关联分析通过WGCNA算法计算各模块基因和样本构成的矩阵(Module Eigengene,ME)与临床性状的皮尔森相关系数及其P值,通过皮尔森相关系数衡量不同模块与临床性状之间的关系,并取相关系数最高的模块用于后续分析。然后利用基因显著性(Gene Significance,GS)和模块显著性(Module Significance,MS)计算与样本种类相关模块的表达模式。GS为每个基因的表达量与某一临床信息的皮尔森相关系数,MS为模块中所有基因的GS取绝对值均值。为了筛选目的模块中的枢纽基因,计算目的模块中每一基因与该模块ME的相关系数,即模块隶属度(module membership,MM),用以衡量目的模块内的某一基因从属于该模块的程度。
1.5 枢纽基因的寻找和分析通过对各个模块和样本临床信息进行关联分析后做出相应热图,选取相关性最高的模块,将该模块中的基因信息导入到Cytoscape(3.7.1版本)软件中,使用Cytoscape将模块中的基因信息可视化为网络,并通过连接度对模块中基因进行排序,筛选出前7个基因用于进一步分析。最后把筛选出的基因用GEPIA数据库做生存分析,观察其对生存预后的影响。
1.6 统计学处理本研究使用R统计软件(3.6版)和WGCNA包进行统计计算。本文采用的食管组织的相关临床特征和各个共表达模块的ME间相关系数的计算是基于R语言平台Rstuio下实现。WGCNA即加权基因共表达网络分析,可被应用于寻找功能相似的基因。WGCNA使用了一种软阈值(soft thresholding)定义一个权重值,判断每一组基因连接的可能性。在此概念下,形成加权共表达网络。WGCNA使用一个以生物学意义作为基准的评判标准——无尺度拓扑学准则。
2 结果
2.1 数据来源及数据预处理从GEO数据库中得到行名为22个样本,列名为33 297个核酸探针的矩阵。利用平台文件GPL6244将探针名转换为基因名,并对其基因表达量作log2处理及标准化处理,最终得到包含个基因的样本基因表达谱矩阵,作为共表达网络构建的输入文件。
2.2 数据筛选本研究选取方差前25%的14 124个基因用于共表达网络的构建。通过构建22个食管组织样本的14 124个基因的层次聚类树(图1),发现无明显离群样本。
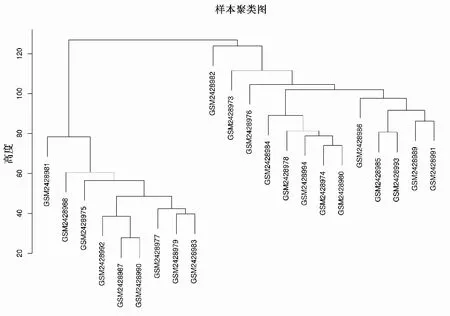
图1 22个食管组织样本系统聚类图
2.3 构建基因共表达网络模块更符合无尺度特征,选取β值为4用于构建共表达网络(图2)。通过动态剪切树法进行模块识别,最终得到45个模块(图3)。随机选取400个基因做TOM热图(图4),描述基因之间的拓扑重叠矩阵,确定每个模块独立存在。
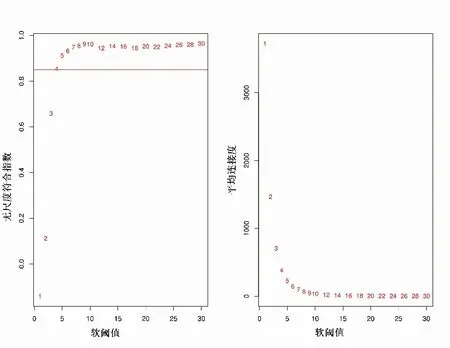
图2 加权基因共表达网络分析软阈值(β)的确定
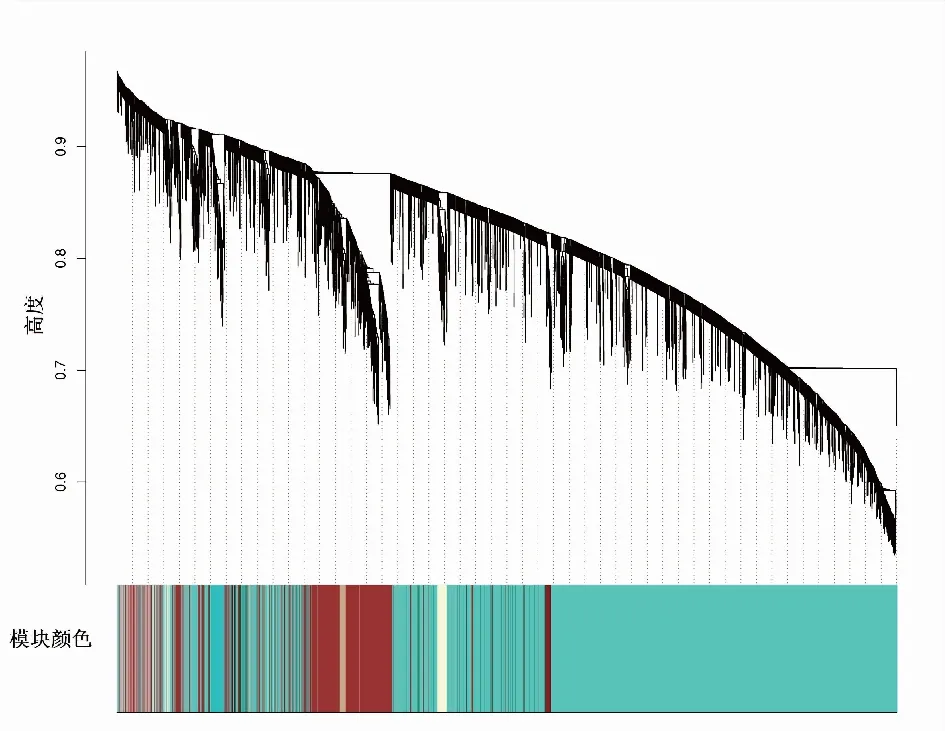
图3 基因聚类树状图
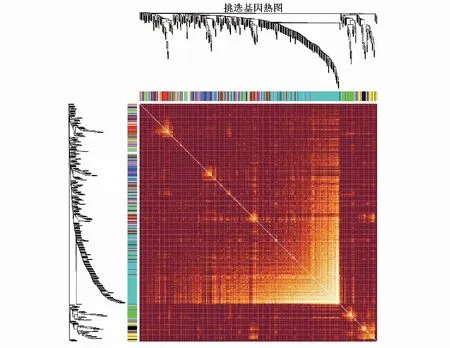
图4 拓扑重叠矩阵(TOM)图
2.4 模块与临床特征关联分析对各个模块和样本临床信息进行关联分析,从模块和性状热图中可以发现深红色(darkred)模块与食管腺癌相关程度最高(图5)。计算深红色模块中基因GS和MM相关系数(cor=0.77,P=5.6e-25)进一步验证此结果的可信度(图6)。结果深红色模块和食管腺癌的关联程度最大,因此后续研究选取深红色模块筛选枢纽基因。
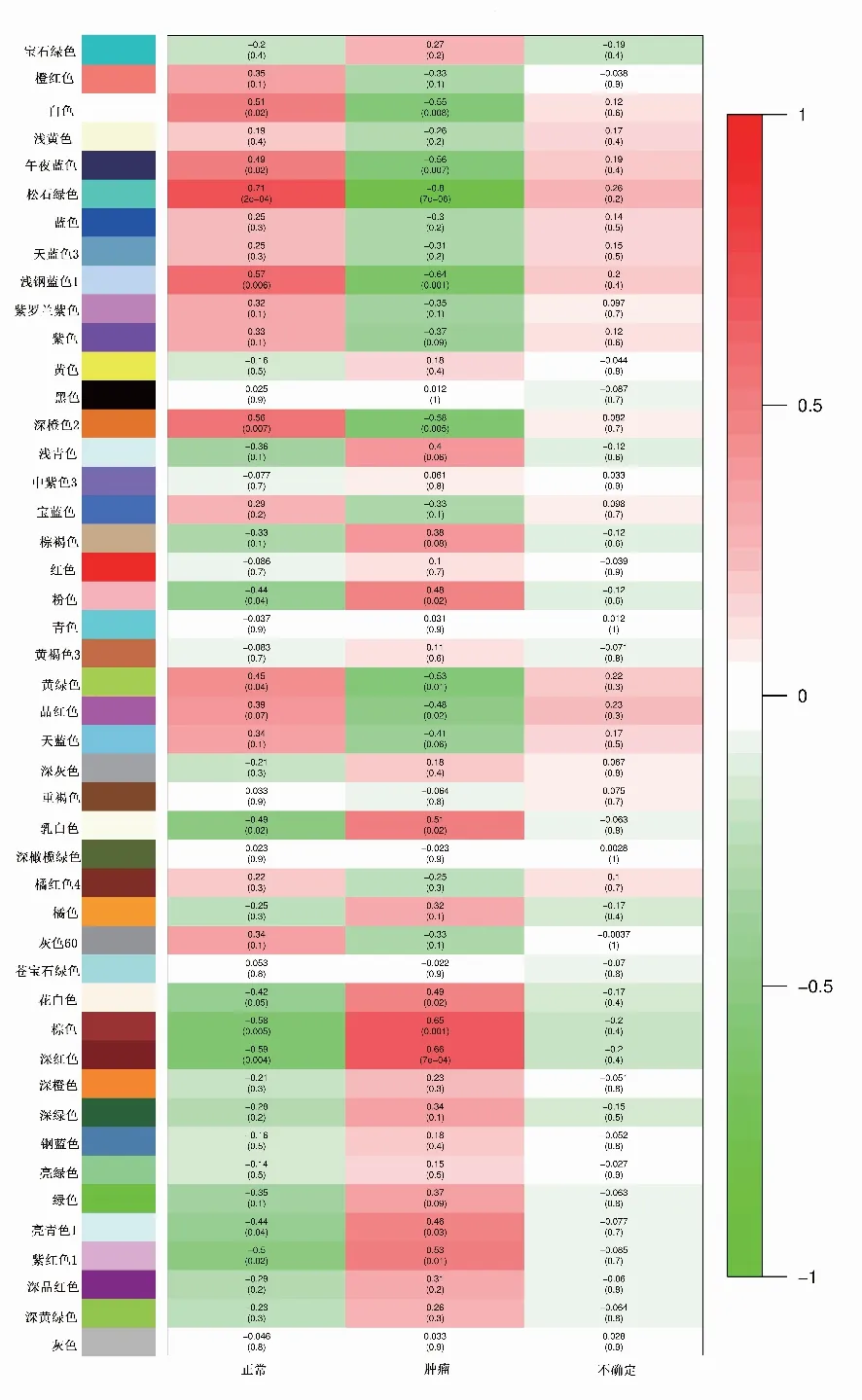
图5 临床特征与模块特征相关性热图
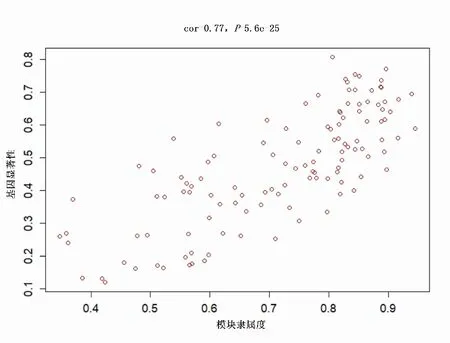
图6 深红色模块基因模块隶属度与基因显著性相关的散点图
图7 矩阵网络热图
2.5 寻找并分析枢纽基因进一步分析发现深红色模块共有121个基因,通过WGCNA对这 121个基因进行连通性分析后导入Cytoscape软件做进一步分析。使用Cytoscape软件对目标模块的网络进行可视化(图8),并依据连接度选出枢纽基因(图9),结果显示深红色模块中的枢纽基因为VASP、BEX4、KDELR3、POLR3B、MYO15B、FBXO27、ABCC3。最后将枢纽基因导入GEPIA数据库中做生存分析(图10)。
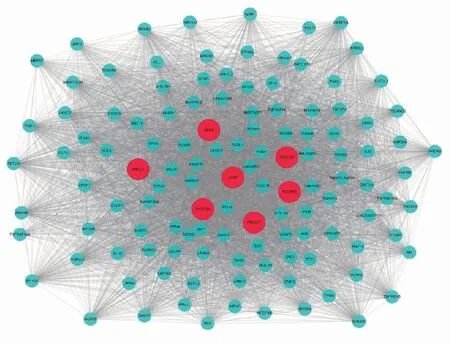
图8 深红色模块中基因网络可视化
3 讨论
食管癌是世界上最为常见的消化系统恶性肿瘤之一,中国食管癌的发病率和病死率居世界第一位[3]。食管癌恶性程度较高,且早期症状不典型,导致早期诊断困难,临床就诊患者多为中晚期,预后极差。随着食管外科手术技术的提高和放射治疗、化学治疗技术的不断创新,食管癌可切除率以及患者生存率有了一定提高,但化学治疗药物毒副作用较大,很多患者无法耐受,且对化学治疗药物有耐药趋势[4]。因此,不断有研究尝试食管癌生物靶向治疗及免疫治疗手段,探索相关分子作用的靶点。传统的基因共表达模式的研究通常通过计算任意两个基因之间的相关系数,设立硬阈值来衡量两个基因是否具有相似表达模式,缺乏说服力。WGCNA通过在高通量数据中识别出功能相关或表达相似的基因构成的模块,从生物功能整体考虑基因功能及其联系,弥补了传统方法的缺陷。通过使用这种方法,研究者不仅可以发现某模块内基因间的联系,还可以关注该模块与其他模块内基因的相关性。此外,通过将临床信息与模块相关联,还可进一步获得与临床特征相关的基因,有助于建立疾病某临床特征的研究基础[5]。
本研究采用WGCNA对GEO数据库中的22个食管组织样本基因表达谱进行分析,最终获得了45个共表达模块。将共表达模块与临床信息相关联,从临床特征与模块特征相关性热图中得到深红色模块中的基因与食管腺癌组织相关性系数最高(0.66,P=7e-04),且与食管正常组织相关性系数最低(-0.59,P=0.004),深红色模块中的基因与食管腺癌的形成高度相关。将深红色模块中121个基因间的连通性信息导入到Cytoscape软件中,进一步识别出7个可能在食管腺癌中发挥关键作用的枢纽基因,分别为VASP、POLR3B、BEX4、KDELR3、ABCC3、FBXO27、MYO15B。VASP作为一种肌动蛋白结合蛋白,参与一系列依赖细胞骨架重塑和细胞极性的过程,如轴突导向、片长轴突和丝状轴突动力学、血小板激活和细胞迁移,VASP与丝状肌动蛋白的形成有关,可能在细胞黏附和运动中起着广泛的作用[6];ABCC3是MDR相关蛋白家族中的一员,能转运谷胱甘肽、硫酸盐、胆汁酸和葡糖苷酸聚合物等多种内源性和外源性化合物,被发现在多个恶性肿瘤中有高表达[7];BEX4通过解除微管动力学和染色体完整性的调控,可抵抗凋亡和细胞死亡,导致非整倍体的获得,同时增加了肿瘤的增殖潜能和生长[8];POLR3B在感应和限制细胞内细菌和DNA病毒感染方面起着关键作用,在先天免疫反应中起核质和胞质DNA传感器的作用[9];KDELR3是编码KDEL内质网蛋白保留受体家族成员,在酵母和动物细胞中,通过从顺式高尔基体或高尔基前腔室中不断获取内质网腔内的可溶性蛋白,可以实现内质网腔内可溶性蛋白的保持[10];FBXO27为SCF (SKP1-CUL1-F-box蛋白)型E3泛素连接酶复合物的底物识别组分,能够识别和结合变性糖蛋白[11]。MYO15B在食管癌及其他肿瘤中的作用及功能尚未见报道,其作用需要进一步验证。
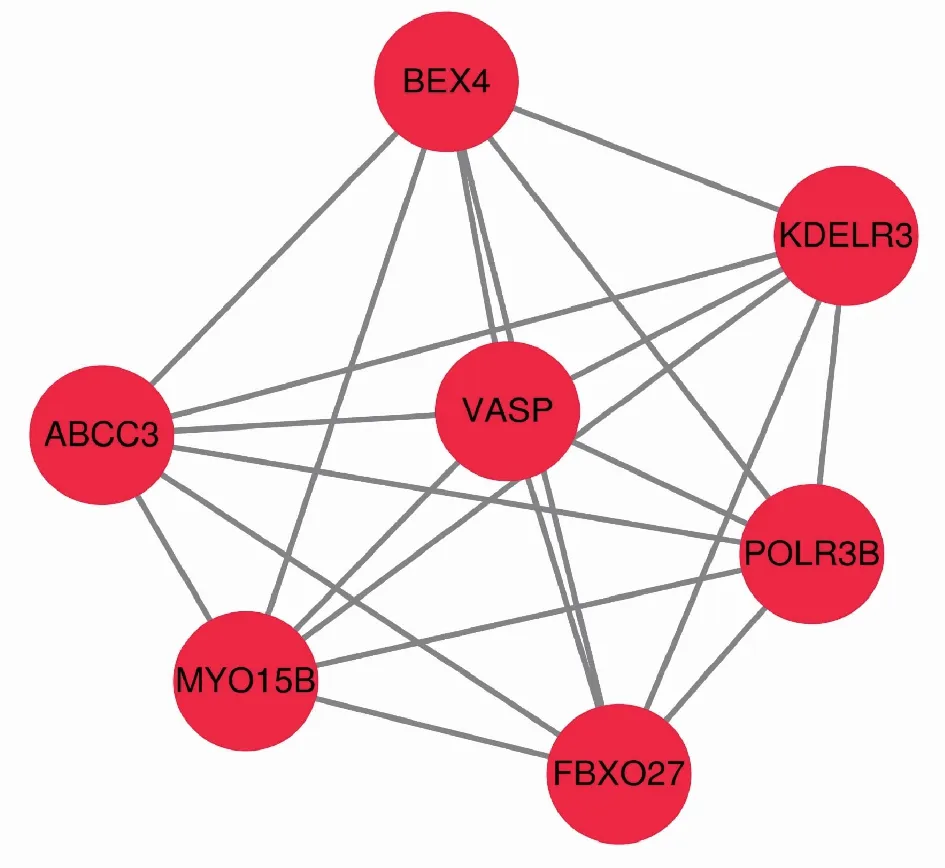
图9 枢纽基因网络可视化
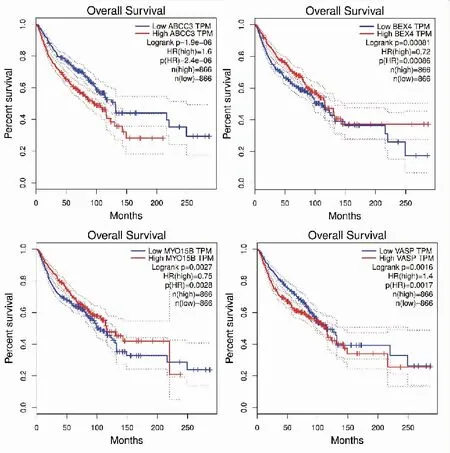
P<0.05,差异有统计学意义。图10 利用GEPIA数据库做枢纽基因的生存分析
将7个枢纽基因导入到GEPIA数据库中做生存分析,发现VASP、ABCC3、MYO15B基因高表达患者总体生存率低于低表达患者,且P<0.05,说明VASP、ABCC3、MYO15B可能为食管腺癌的致癌基因。BEX4基因高表达患者总体生存率高于低表达患者,BEX4可能为食管腺癌的抑癌基因。因此这些基因可能是影响食管腺癌生存和预后的关键基因,可能作为食管腺癌诊断和治疗的潜在靶点,对进一步理解食管腺癌的分子机制可起到一定帮助。需要进一步对这些基因的作用进行单独的分析和实验验证。
