CK2介导幽门螺杆菌CagA 蛋白诱导人胃腺癌上皮细胞内IL-8表达的机制研究△
2019-12-23顾岚陈颖周仁民林琼
顾岚,陈颖,周仁民,林琼
无锡市儿童医院消化科,江苏 无锡 214000
幽门螺杆菌(helicobacter pylori,Hp)是一种革兰阴性螺杆菌,主要在儿童时期经粪-口、口-口、胃-口等途径感染,感染后可引起胃黏膜急慢性炎症、消化性溃疡、胃癌等疾病[1-3]。细胞毒素相关基因A(cytotoxin associated gene A,CagA)编码的CagA蛋白是Hp重要的毒力因素之一,而白细胞介素-8(interleukin-8,IL-8)作为Hp感染后急性胃炎的重要调节因子,与CagA的关系较为密切[4-6]。核因子κB(nuclear factor-κB,NF-κB)参与机体的炎症反应过程,Hp感染可促进NF-κB活化,进而促进多种炎症因子的表达,包括IL-8[7-8]。研究表明,多种激酶可调节NF-κB p65的磷酸化,包括蛋白激酶CK2[9-10]。CK2是一种多功能信使非依赖性丝氨酸/苏氨酸蛋白激酶,可参与细胞增殖、癌基因调节及信号转导,与自身免疫性疾病、病毒及寄生虫感染和各种恶性肿瘤有关[11-13]。许多细胞系研究中已经证实,CK2与NF-κB激活及趋化因子表达有关[10-13]。本研究探讨了CK2介导CagA蛋白诱导人胃腺癌上皮细胞(human gastric adenocarcinoma cell,AGS)内IL-8表达的可能机制,现报道如下。
1 材料与方法
1.1 材料与试剂
人AGS细胞购自中国科学院上海生命科学研究院细胞资源中心,F12K培养基购自美国赛默飞世尔科技公司,胎牛血清、双抗(青霉素/链霉素)混合液均购自美国Gibco公司,重组Hp CagA蛋白、p65、p65-p ser529兔单克隆抗体均购自英国Abcam公司,芹菜素(apigenin)购自英国Tocris公司,人IL-8酶联免疫吸附测定(enzyme-linked immunosorbent assay,ELISA)试剂盒购自美国R&D公司,p65siRNA购自美国SantaCruz公司,β-肌动蛋白(β-actin)购自美国CST公司,山羊抗兔二抗购自美国Jackson公司;聚偏二氟乙烯(polyvinylidene fluoride,PVDF)膜、电化学发光(electrochemiluminescence,ECL)液购自美国Millipore公司,Lipofectamine2000购自美国Invitrogen公司,RIPA裂解液、5×蛋白上样缓冲液、pNF-kB-luc质粒、pRLSV40-C质粒及双萤光素酶报告基因检测试剂盒均购自上海碧云天生物技术有限公司。
1.2 细胞培养和处理
将人AGS细胞置于含10%胎牛血清和1%双抗的F12K培养基中,于5%CO2、37℃恒温培养箱中培养,取3~6代细胞进行实验。根据不同实验条件分组如下。S1组加入CagA,S2组未加入CagA[加入磷酸盐缓冲液(phosphate buffered saline,PBS)作为空白对照],实验条件:将AGS细胞内分别加入CagA、PBS孵育24 h。A1组未转染p65siRNA且未加入CagA,A2组转染p65siRNA但未加入CagA,A3组未转染p65siRNA但加入CagA,A4组转染p65siRNA且加入CagA,实验条件:AGS细胞转染或不转染p65siRNA 24 h后,再加入或不加入CagA蛋白孵育24 h。A组转染ConsiRAN但未转染p65siRNA,B组转染p65siRNA但未转染ConsiRAN,实验条件:AGS细胞转染p65siRNA或ConsiRAN 24 h。B1组未加入apigenin(CK2β抑制剂)和CagA,B2组加入apigenin但未加入CagA,B3组未加入apigenin但加入CagA,B4组加入apigenin和CagA,实验条件:AGS细胞中加入或不加入apigenin 30 min后,再加入或不加入CagA蛋白孵育24 h。C组为加入CagA 0 min时,D组为加入CagA 20 min时,实验条件:AGS细胞内加入CagA蛋白0、20 min。上述所有分组中,CagA蛋白浓度为10 μg/ml,p65siRNA 浓度为 50 nmol/L,ConsiRNA浓度为 50 nmol/L,apigenin 浓度为 10 μmol/L。所有空白对照均为PBS。
1.3 蛋白质印迹法(Western blot)检测p65-p、p65蛋白表达
采用Western blot检测不同条件下AGS细胞中p65-p ser529、p65、β-actin的表达情况。采用PBS洗涤细胞2次后,4℃下将细胞置于RIPA裂解液中裂解30 min,然后4℃ 15 000 r/min,离心20 min。采用二喹啉甲酸(bicinchoninic acid,BCA)蛋白定量试剂盒检测蛋白浓度。取40 μg蛋白进行十二烷基硫酸钠-聚丙烯酰胺凝胶(sodium dodecyl sulfate-polyacrylamide gelelectrophoresis,SDS-PAGE)电泳,PVDF转膜后放入特异性抗体中4℃孵育过夜,然后采用辣根过氧化物酶标记的二抗共孵育1 h,ECL法显示结果,凝胶成像系统扫描,根据条带灰度显示情况判断表达情况。
1.4 ELISA 检测
采用ELISA试剂盒测定在不同实验条件下AGS细胞质IL-8蛋白的表达水平。
1.5 双荧光素酶报告系统检测NF-κB转录活性
将细胞接种于24孔板中,2 h后换为Opti-MEN培养基。使用Lipofectamine2000将pNF-kB-luc与pRL-SV40-C按10∶1比例混合后转染至AGS细胞。转染后继续培养6 h后,更换为F12K完全培养基继续培养18 h。根据实验分组在加入apigenin(10 μmol/L)30 min后再加入CagA(10 μg/ml)蛋白继续培养24 h。去除培养液,PBS洗涤,加入RIPA裂解液后充分裂解细胞,12 000g离心15 min,取上清液后置于96孔板中,按序加入萤火虫荧光素酶检测试剂和海肾萤光素酶检测试剂,以空白孔为参照测定每孔的相对荧光信号。实验重复3次,每次设3个复孔。相对荧光强度=(萤火虫荧光素酶RLu实验组/海肾荧光素酶RLu实验组)/(萤火虫荧光素酶RLu对照组/海肾荧光素酶RLu对照组)。RLu为相对荧光素酶活性。
1.6 统计学分析
采用Excel 2010及GraphPadPrism 5软件进行统计分析;计量资料以均数±标准差(±s)表示,组间比较采用t检验。以P<0.05为差异有统计学意义。
2 结果
2.1 CagA诱导AGS细胞释放IL-8
S1组AGS细胞中IL-8蛋白的表达水平为(871.25±12.89)pg/ml,高于S2组的(119.00±12.70)pg/ml,差异有统计学意义(t=83.125,P<0.05)。
2.2 NF-κB p65参与CagA诱导AGS细胞表达IL-8
B组细胞的p65蛋白表达水平低于A组(图1)。A1组和A2组细胞的IL-8蛋白表达水平比较,差异无统计学意义(P>0.05);A4组细胞的IL-8蛋白表达水平明显低于A3组,差异有统计学意义(t=72.976,P<0.01)(表1)。

图1 Western blot 检测不同组别AGS细胞中p65蛋白表达
表1 不同组别(p65 siRNA、CagA转染或加入情况不同)AGS细胞中IL-8蛋白的表达水平(pg/ml,±s)

表1 不同组别(p65 siRNA、CagA转染或加入情况不同)AGS细胞中IL-8蛋白的表达水平(pg/ml,±s)
组别IL-8蛋白表达水平A1组A2组A3组A4组668.25±11.09 656.25±5.91 1785.50±20.70 907.00±12.30
2.3 CK2参与CagA诱导AGS细胞表达IL-8
B1组和B2组细胞的IL-8蛋白表达水平比较,差异无统计学意义(P>0.05);B4组细胞的IL-8蛋白表达水平明显低于B3组,差异有统计学意义(t=15.911,P<0.01)。(表2)
2.4 CK2介导CagA诱导的AGS细胞内NF-κB p65磷酸化及转录激活
D组细胞的p65-p蛋白表达水平高于C组(图2)。B4组细胞的p65-p蛋白表达水平低于B3组(图3)。B1组和B2组细胞的相对荧光强度比较,差异无统计学意义(P>0.05);B4组细胞的相对荧光强度明显低于B3组,差异有统计学意义(χ2=25.149,P<0.01)(表3)。
表2 不同组别(apigenin和CagA加入情况不同)AGS细胞中IL-8蛋白的表达水平(pg/ml,±s)

表2 不同组别(apigenin和CagA加入情况不同)AGS细胞中IL-8蛋白的表达水平(pg/ml,±s)
组别IL-8蛋白表达水平B1组B2组B3组B4组52.75±7.18 45.00±6.22 893.75±37.72 469.75±37.65

图2 Western blot 检测不同组别(CagA 加入时间不同)p65-p蛋白表达
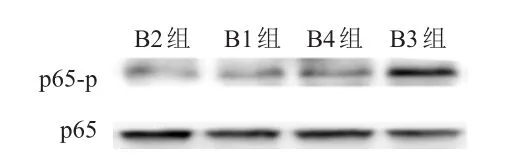
图3 Western blot 检测不同组别(CagA和apigenin加入情况不同)p65-p蛋白表达
表3 不同组别(CagA和apigenin加入情况不同)AGS细胞的相对荧光强度(±s)

表3 不同组别(CagA和apigenin加入情况不同)AGS细胞的相对荧光强度(±s)
组别 相对荧光强度B1组B2组B3组B4组1.00±0.06 0.95±0.04 2.63±0.06 1.47±0.07
3 讨论
Hp感染可引起急慢性胃炎、消化性溃疡、胃癌、黏膜相关淋巴瘤等,不同的转归状态与多种因素有关,CagA是重要因素之一[14]。CagA基因编码的CagA蛋白与Hp感染导致的胃黏膜损伤程度和临床症状的严重程度相关[15]。CagA蛋白可以促进NF-κB激活,诱导炎症介质基因的转录表达(包括IL-8)。CagA蛋白能够诱导胃黏膜上皮细胞分泌IL-8,促进嗜中性粒细胞趋化至胃黏膜部位,导致胃黏膜产生较强烈的炎症反应[4]。研究发现,CagA阳性菌株感染的AGS细胞中IL-8的表达水平高于CagA阴性菌株感染的细胞[16]。本研究中,CagA蛋白刺激AGS细胞后,IL-8的表达水平升高,与上述研究结果一致。
NF-κB是一种重要的多功能核转录因子,参与多种基因的转录调控,调节机体感染后的炎症反应过程。在非活化状态下,NF-κB在细胞质内与κB 抑制蛋白(inhibitor of κB,IκB)结合,IκB 激酶被激活后导致IκB蛋白被降解,NF-κB得到释放,随后NF-κB被进一步激活并转移至细胞核内与目的基因结合并促进其转录表达[17]。研究表明,Hp感染可以引起AGS细胞或胃癌患者胃黏膜中NF-κB活化,促进多种炎症因子的表达,如IL-8、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、白细胞介素-1(interleukin-1,IL-1)[18]。研究表明,AGS 细胞或患儿胃黏膜感染Hp后,CagA阳性菌株较CagA阴性菌株更能促进局部NF-κB的大量激活[18]。本研究证实,p65siRNA能够沉默AGS细胞中p65的表达,进一步实验发现,应用CagA蛋白刺激后,IL-8表达水平升高,但在p65siRNA沉默的细胞中IL-8表达水平较低。同时,本研究发现,与未加入CagA蛋白比较,CagA蛋白刺激后,NF-κB p65在ser529位点的磷酸化水平增加,表明NF-κB p65参与CagA诱导AGS细胞表达IL-8过程。
NF-κB亚基p65的磷酸化可增强NF-κB的转录激活能力及其与p300的结合能力,从而诱导目的基因的表达。研究表明,CK2可调节NF-κB p65磷酸化[9-10]。CK2是一种多功能信使非依赖性丝氨酸/苏氨酸蛋白激酶,由两个催化亚基(α和α')和两个调节亚基(β)组成,参与多种病理过程[7]。研究表明,CK2能够促进p65在ser529位点的磷酸化,而抑制这一过程后p65的转录活性下降[19]。本研究中,CagA蛋白刺激AGS细胞后IL-8表达增加,但使用apigenin抑制CK2β表达后,再使用CagA蛋白刺激AGS细胞,AGS细胞内IL-8表达降低,表明CK2参与CagA诱导AGS细胞表达IL-8的过程。进一步实验发现,与加入CagA但未加入apigenin蛋白的AGS细胞相比,同时加入apigenin和CagA蛋白的AGS细胞中,p65在ser529位点的磷酸化水平下降,这表明CK2介导CagA诱导的NF-κB p65在ser529位点的磷酸化过程。本研究也检测了NF-κB的转录活性,同时加入apigenin和CagA蛋白组中AGS细胞内荧光素酶活性较加入CagA但未加入apigenin蛋白组显著下降,表明CK2介导CagA诱导的AGS细胞内NF-κB的转录激活。
综上所述,CK2可能参与了CagA诱导AGS细胞中NF-κB激活及IL-8表达的过程。
