甘南地区牦牛曲拉中细菌群落结构
2019-12-04梁春御曹瑛瑛文开勇文鹏程冯晓蘶张忠明张卫兵
曹 磊,梁春御,曹瑛瑛,文开勇,文鹏程,杨 敏,冯晓蘶,张忠明,*,张卫兵
(1.甘肃农业大学食品科学与工程学院,甘肃 兰州 730070;2.兰州资源环境职业技术学院气象学院,甘肃 兰州 730021;3.甘肃食品药品监督管理局,甘肃 兰州 730070;4.甘肃农业大学理学院,甘肃 兰州 730070)
牦牛曲拉,又称奶渣,是将牦牛乳脱脂后,在自然条件下进行发酵使酪蛋白凝结、干燥后制成的一种发酵乳制品[1]。与新鲜牦牛乳比较,曲拉具有蛋白质含量较高、易贮藏、方便运输的特点[1-2]。曲拉不仅是藏区牧民贮藏食品蛋白的重要手段,还是制作酸奶的发酵剂。另外,曲拉还可作为生产干酪素的原料[3]。由于曲拉加工环境相对开放,因此环境中的多种微生物参与了发酵过程[4]。
目前,微生物多样性的分析方法主要包括传统纯培养方法和基于分子生物学技术的非纯培养的方法。纯培养的方法只能在实验室条件下对可培养的微生物进行分析,不能全面了解微生物多样性;非纯培养的方法包括变性梯度凝胶电泳[5]、单链构象多态性[6]、温度梯度凝胶电泳[7]、末端限制性片段长度多态性分析[8-10]等,其早期也广泛应用于乳制品中微生物多样性的研究[11-13]。然而,近年来新兴的Illumina MiSeq高通量测序技术能够全面准确地对研究对象中微生物种类组成和结构进行分析,相较于传统的纯培养方法及以16S rRNA为基础的非纯培养方法,能够较多的产生测序覆盖深度更大的数据量,更全面的呈现其微生物多样性,明确各类微生物的结构比例及优势菌群。该技术已在原乳[14]、发酵酸牛奶[9]、发酵酸驼奶和发酵酸马奶[15]的微生物多样性研究中得到了成功应用。
本研究采用Illumina MiSeq高通量测序技术对采自于甘肃省甘南藏族自治州的曲拉样品中细菌多样性进行分析,以期全面地解析曲拉中的细菌多样性,系统地了解曲拉中细菌组成及群落结构。
1 材料与方法
1.1 材料与试剂
曲拉样品采自于甘肃省甘南藏族自治州合作市那吾乡玛岗村(MG1、MG2、MG3)、卡斯合村(KS1、KS2、KS3)、绍玛村(SM1、SM2、SM3)3 个村庄的9 个不同牧民家庭。将所有样品置于自封袋中编号,并置于冰盒中运输至实验室以备实验。
E.Z.N.A.Soil DNA试剂盒 美国Omega公司;Qubit 2.0 DNA检测试剂盒 美国Invitrogen公司;Q5高保真DNA聚合酶 美国New England Biolabs公司;凝胶回收试剂盒 美国Axygen公司;TruSeq Nano DNA LT Library Prep Kit 美国Illumina公司。
1.2 仪器与设备
Pico-21型台式离心机 美国Thermo Fisher科技公司;DYY-6C型电泳仪、DYCZ-21型电泳槽 北京市六一仪器厂;凝胶成像系统 美国UVP公司;Q32866型Qubit 2.0分光光度计 美国Invitrogen公司;T100TMThermal Cyeler型聚合酶链式反应(polymerase chain reaction,PCR)仪 伯乐生命医学产品(上海)有限公司;MiSeq System SY-410-1003高通量测序仪 美国Illumina公司。
1.3 方法
1.3.1 曲拉微生物总DNA的提取
取0.2 g曲拉样品,采用E.Z.N.A.Soil DNA Kit D5625-01提取试剂盒,参照说明书从样品中提取DNA;然后使用Qubit 2.0分光光度计对提取的DNA进行定量,并通过0.8%琼脂糖凝胶电泳确定DNA提取的完整性。
1.3.2 PCR扩增及测序
以稀释后的基因组总DNA为模板,取30 ng进行PCR靶向扩增16S rRNA基因。采用V3-V4区338F(5’-ACTCCTACGGGAGGCAGCAG-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’)分别为特异性引物。16S rRNA基因PCR热循环谱如下:95 ℃预变性5 min;95 ℃变性30 s,56 ℃退火30 s,72 ℃延伸30 s,25 个循环;72 ℃退火10 min,最终保持在4 ℃条件下。PCR产物使用2%琼脂糖凝胶电泳进行检测,切割后使用Axygen公司的凝胶回收试剂盒回收。将扩增产物纯化后,利用分光光度计精确定量后制备测序文库,最后在MiSeq测序平台进行双端测序。PCR扩增、测序文库制备及测序工作由上海派森诺生物科技股份有限公司完成。
1.3.3 高通量测序数据处理
采用Mothur(version 1.31.2)和QIIME(version 1.7.0)软件处理及分析高通量测序数据[16-17]。先对测序结果进行整合,使每次读数的碱基平均质量不小于Q20。然后将整合后的序列使用FLASH(version 1.2.7)软件根据重叠碱基进行配对连接,并丢弃无法配对的序列[18]。使用UCHIME(version 4.2)软件鉴定和去除嵌合体序列[19-20]。将序列分配入对应样本,使用UCLUST算法进行序列聚类,以97%的序列相似度作为划分阈值,将样品聚类成可操作分类单位(operational taxonomic units,OTU)[18]。
1.3.4 群落多样性和统计分析
利用Mothur(version 1.31.2)软件进行α多样性分析,其中包括Simpson指数、Chao1指数和Shannon指数,并在不同的分类水平上对群落结构进行了统计分析。
使用QIIME软件对基于Unweighted和Weighted的UniFrac距离矩阵进行UPGMA聚类分析,并使用R(version 2.15.3)软件进行可视化。
通过PICRUSt(version 1.0)软件对群落基因功能特征进行预测[21]。
1.4 数据分析
利用Excel 2007软件进行数据处理分析并作图。
2 结果与分析
2.1 PCR扩增、测序序列及OTU基本情况
由图1可以看出,PCR扩增后得到清晰明显的条带,可用于后续的研究分析。通过细菌的16S rRNA基因V3-V4区测序,9 份样品共产生216 630 条有效序列,将得到的有效序列经过质量控制,在剔除一些疑问序列之后得到高质量序列为185 910 条,高质量序列占有效序列的比例为85.82%。将所有序列按97%的相似度进行OTU聚类,得到4 754 个OTU。由序列数及OTU聚类可以看出,曲拉中细菌种类繁多,物种丰富,且不同来源的曲拉样品中存在一定差异。

图1 PCR扩增产物凝胶电泳图Fig. 1 Electrophoresis profile of PCR amplified product
2.2 稀疏曲线
稀疏曲线用来评判每个样品在当前的测序深度下是否足以反映该样品中所包含的细菌群落多样性。由图2可以看出,当样品测序量较低时,每个样品的Shannon指数呈显著上升的趋势,说明在当前测序量上样品物种的多样性较高,样品中还有较多的物种没有被检测到。但随着测序数量逐渐增加,各曲拉样品中Shannon指数呈缓慢增加的趋势,稀疏曲线的斜率逐渐降低,并且随着测序数量的继续增加,Shannon曲线基本稳定。虽然在当前的测序深度下每个样品的稀疏曲线未能完全进入平台期,但与X轴几乎接近平行,已经达到饱和。因此,尽管随着测序量的增加新的物种可能会被发现,但在此测序水平下,样品中细菌的群落多样性已能够充分的展现。

图2 Shannon指数稀疏分析图Fig. 2 Sparse analysis of Shannon index
2.3 α多样性分析
曲拉样品中微生物α多样性指数如表1所示。对于微生物群落而言,可以用Chao1指数、Shannon指数、ACE指数和Simpson指数反映样品中物种的丰富度和多样性。由表1可以看出,曲拉样本中细菌的Chao1指数平均值为307.06±124.62,ACE指数为473.93±180.14,Simpson指数为0.74±0.13,Shannon指数为3.29±1.35。从微生物多样性指数结果分析可以得出,曲拉样品中细菌群落的物种多样性及总体丰度相对较高,曲拉中的微生物数量处于较高的水平。同时,不同村庄来源的样品微生物多样性指数存在差异,这表明样品制备环境可能会影响样品中的微生物物种丰富度及多样性。Matsoni[22]和Tarag[23]对于发酵乳的研究中也发现了相同的结果。在该研究中所有样品的覆盖率范围为99.91%~99.98%,均大于99%,这表明测序水平能够达到鉴定样品中的大多数微生物群落的多样性。
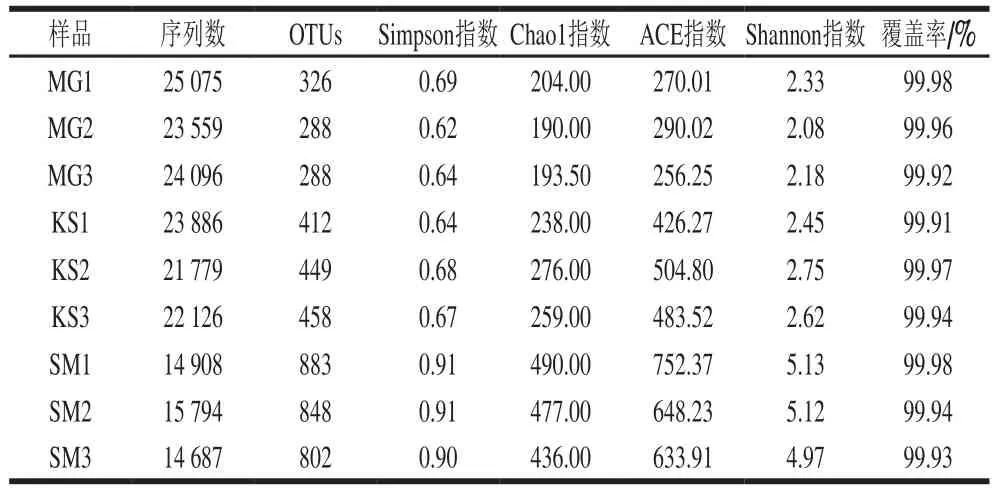
表1 微生物多样性指数Table 1 Microbial diversity indexes
2.4 样品细菌群落在门水平的分类及比较
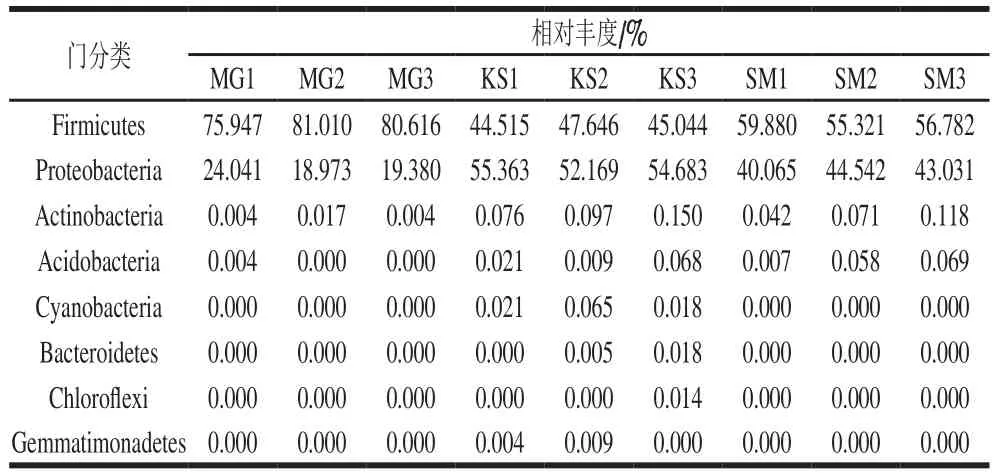
表2 各样品门水平菌群分布相对丰度Table 2 Relative abundance of bacterial flora distribution in Qula samples at phylum level
对9 份曲拉样品从门的分类水平进行鉴定,结果如表2所示。在曲拉样品中共检测出8 个门,分属于厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、蓝藻门(Cyanobacteria)、拟杆菌门(Bacteroidetes)、绿弯菌门(Chlorof l exi)和假单胞菌门(Gemmatimonadetes)。由表2可知,厚壁菌门为曲拉样品中的第1优势门,其相对丰度在44.515%~81.010%的范围内,平均相对丰度为60.751%;其次为变形菌门,为样品中的第2优势门,其相对丰度介于18.973%~55.363%之间,平均相对丰度为39.138%;放线菌门和酸杆菌门丰度较低,平均相对丰度仅为0.064%和0.026%,为样品中的非优势门;蓝藻门(平均相对丰度为0.012%)、拟杆菌门(平均相对丰度为0.003%)、绿弯菌门和假单胞菌门(平均相对丰度为0.001%)的丰度都很低,为曲拉样品中的稀有门。厚壁菌门和变形菌门也是牛乳、马乳、驼乳及其发酵酸乳中的优势门[15],与本实验的研究结果一致。另外,在发酵牛乳[24]、发酵牦牛乳[25]、发酵马乳[15,26]中均检测到绿弯菌门、酸杆菌门、蓝藻门,且均为非优势门,而在先前的研究中没有检测到假单胞菌门。
2.5 样品细菌群落在属水平的分类及比较
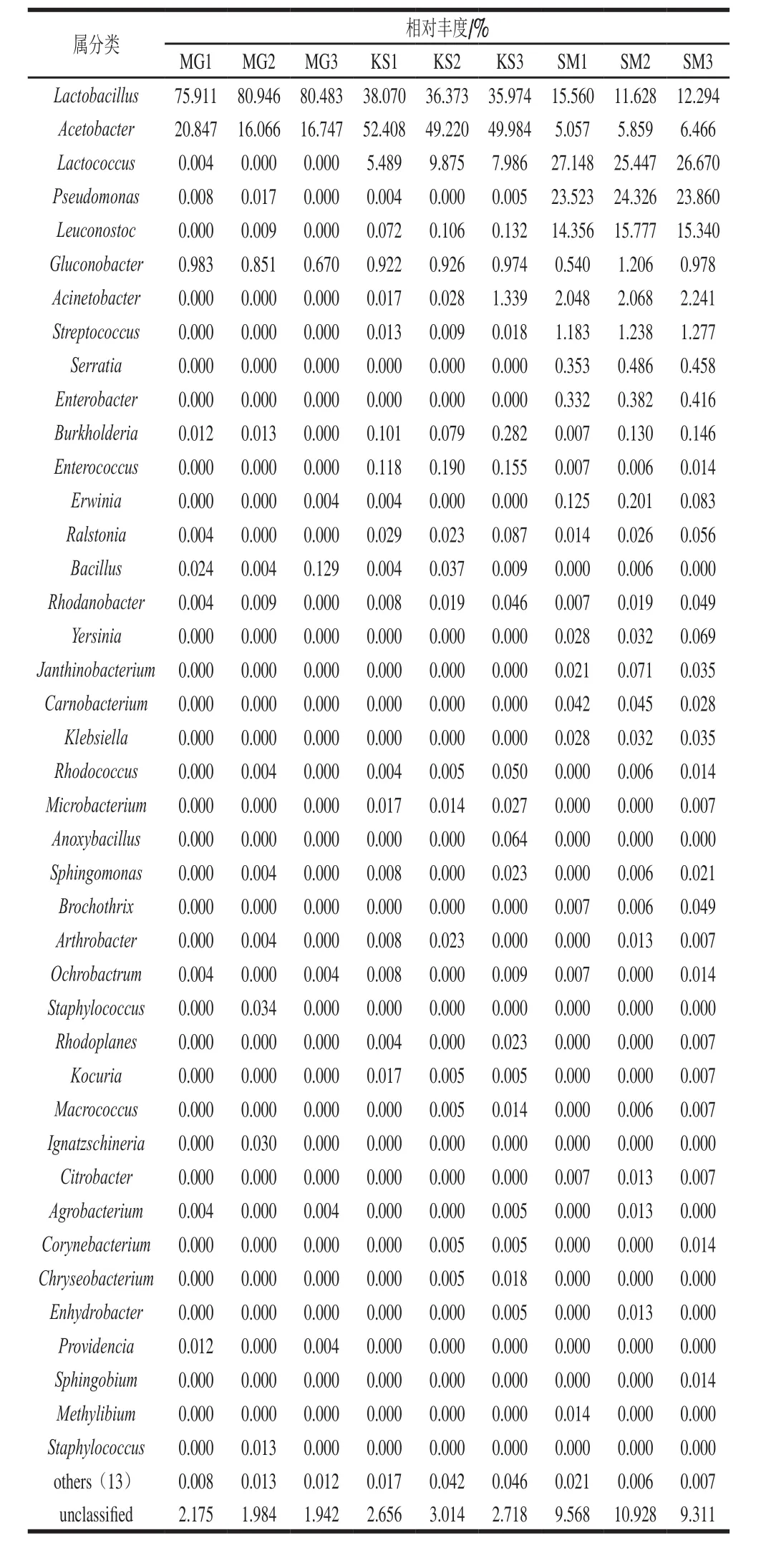
表3 各样品属水平菌群分布相对丰度Table 3 Relative abundance of bacterial flora distribution in Qula sample at genus level
对9 份曲拉样品从属的分类水平进行鉴定,共检测出55 个属,结果如表3所示。从属分类水平来看,在曲拉样品中检测出的乳杆菌属(Lactobacillus)、醋酸杆菌属(Acetobacter)、乳球菌属(Lactococcus)、假单胞菌属(Pseudomonas)和明串珠菌属(Leuconostoc)为丰度相对较高的属,其相对丰度均大于5%。厚壁菌门的乳杆菌属的相对丰度在11.628%~80.946%的范围内,平均相对丰度为43.027%,为曲拉样品中的第1优势属;变形菌门的醋酸杆菌属的相对丰度在5.057%~52.408%的范围内,平均相对丰度为24.739%,为第2优势属;厚壁菌门的乳球菌属的相对丰度相对较高,平均相对丰度为11.402%,为第3优势属。假单胞菌属和明串珠菌属的平均相对丰度分别为7.971%和5.088%。不同来源的曲拉样品在属水平上细菌组成存在差异。另外,样品中还包含许多在属水平上未鉴定的属(unclassified),未鉴定出属也具有较高的丰度,其相对丰度在1.942%~10.928%的范围内,平均相对丰度为4.922%。
肠球菌属(Enterococcus)和葡萄球菌属(Staphylococcus)为样品中的低丰度属,其平均相对丰度分别为0.054%和0.001%。肠球菌属及葡萄球菌属属于食源性病原菌属,说明曲拉相对粗放的生产环境有一定的影响。
不同乳制品的微生物组成有着丰富的多样性,如传统酸奶[23]、发酵牛乳[24,26-27]、发酵马乳[26]、发酵牦牛乳[25]和开非尔[28]的优势属多为乳杆菌属,与本实验的研究结果一致。奶酪中的优势属各不相同,爱尔兰奶酪[29]为乳球菌属,丹麦原奶酪[28]中优势菌株归属于乳杆菌属和链球菌属。有些发酵乳制品样品中也检测到埃希氏杆菌属(Escherichia)和沙门氏菌属(Salmonella)[23],葡萄球菌属和志贺氏菌属(Shellogella)[14]等也有检测到。
2.6 物种丰度热图
丰度热图颜色越绿则说明所含的菌属较少。由图3可以看出,9 个样本的前5 种菌属的显示颜色比较红,含量较多,分别是乳杆菌属、醋酸杆菌属、乳球菌属、假单胞菌属、明串珠菌属。另外,一些没有与数据库匹配的未鉴定出属也具有较高的丰度。不同来源的曲拉样品菌属中有的样品颜色比较红,有的样品颜色比较绿,说明每个样品中菌群的丰度存在差异,因此,不同来源的样品中菌群多样性存在差异性。
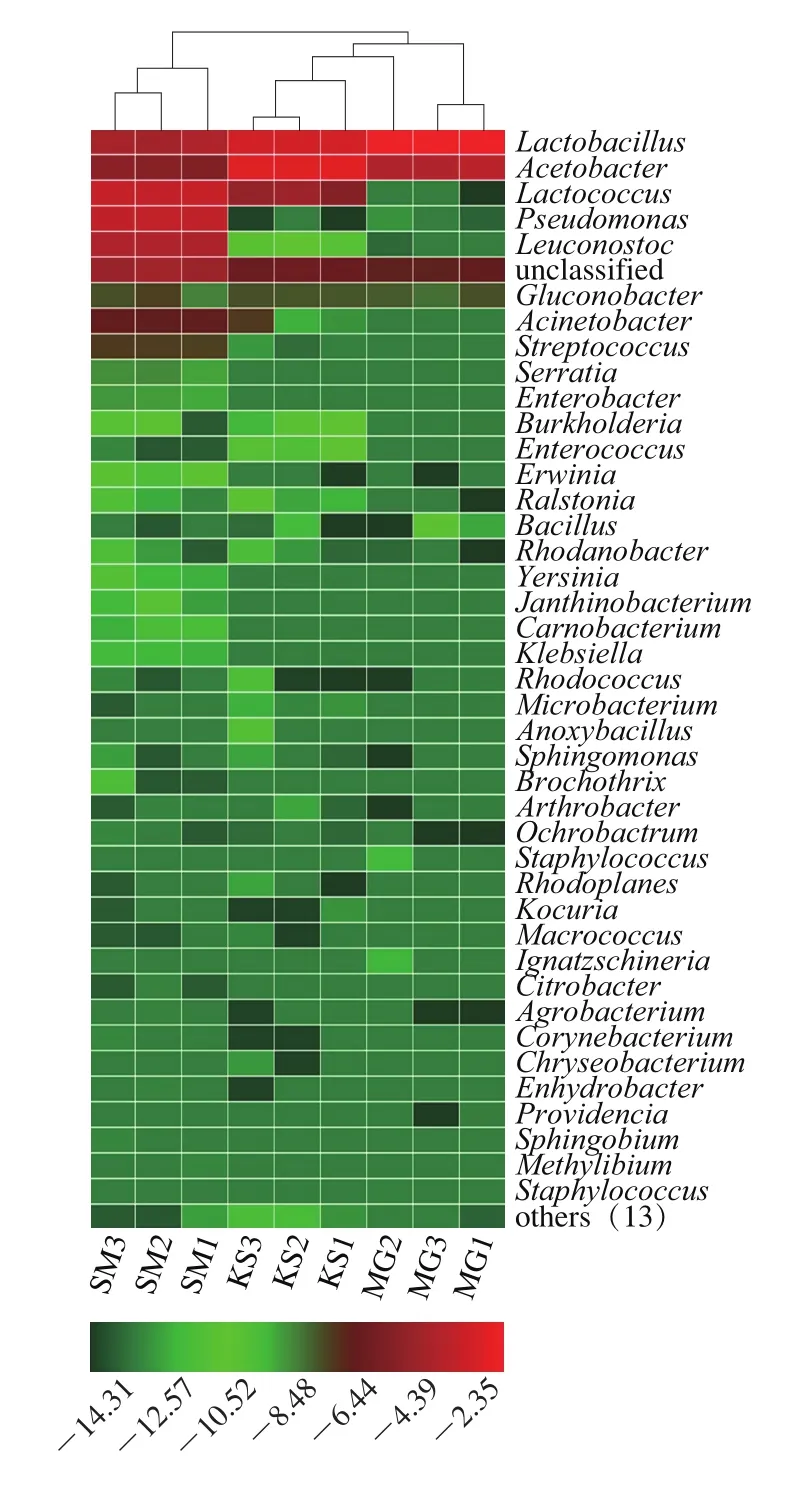
图3 属水平物种丰度热图Fig. 3 Heatmap of species abundance at genus level
2.7 基于UniFrac距离的样本聚类分析
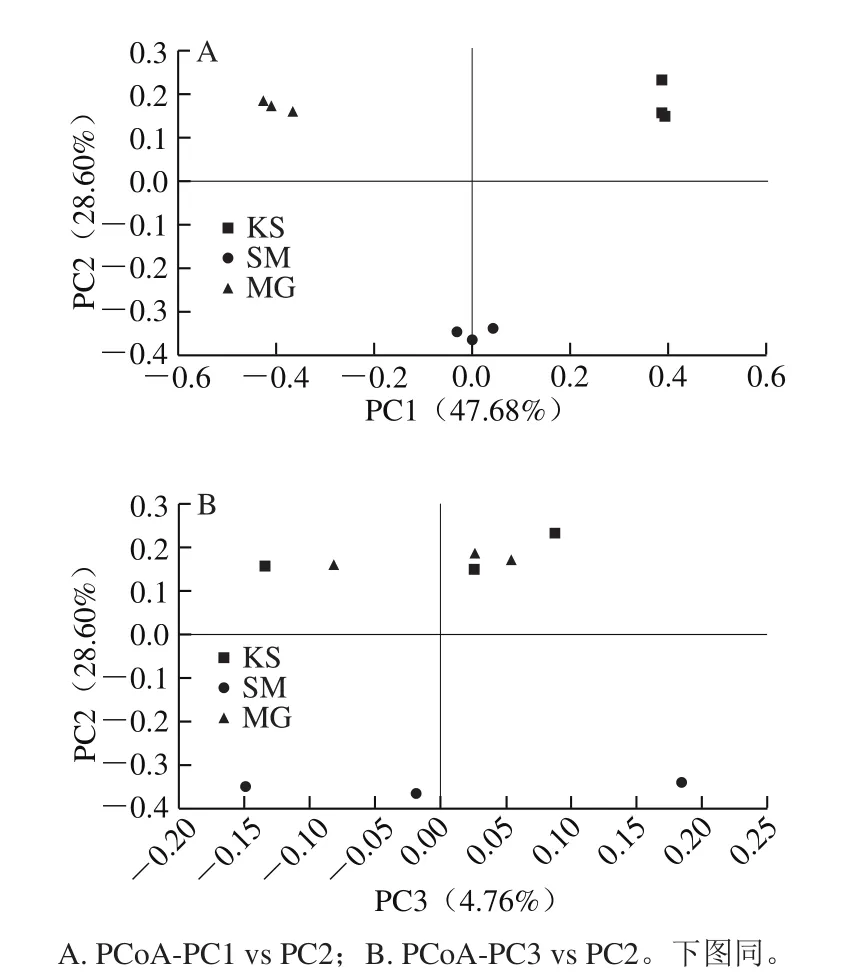
图4 曲拉中菌群结构UniFrac非加权主坐标分析Fig. 4 Unweighted UniFrac principal coordinate analysis of bacterial communities in Qula samples
采用基于UniFrac距离的Weighted和Unweighted主坐标分析对样品中细菌群落结构进行比较,结果分别见图4、5。图4为基于非加权UniFrac距离的主坐标分析,其第1主成分、第2主成分和第3主成分的贡献率分别为47.68%、28.60%和4.76%,不同来源的样品在PC1和PC2维度上明显的呈聚集和分离趋势,个别样品之间存在部分交叠,能够将不同样品较好的区分,而在PC3维度上所有样品可以达到很好区分效果。
图5为基于加权UniFrac距离的主坐标分析,其第1主成分、第2主成分和第3主成分的贡献率分别是71.75%、27.93%和0.15%,不同样品在PC1和PC2维度上有样品之间存在交叠现象,而在PC3维度上所有样品可以很好地区分。
由样品主坐标分析可知,所有样品没有紧密地组合在一起,各样品所代表的点在空间分布有一定的距离,说明不同位置来源的曲拉样品的细菌群落组成具有差异性。本研究中使用的曲拉样品是通过传统方法生产的,因此,样品间细菌群落结构的差异不同很可能归因于原料、地理位置和其他环境因素。从图6可以看出,不同采样地的样品聚集到不同的类别,说明不同来源样品的微生物多样性存在一定的差异性,这与主坐标分析的结果一致。
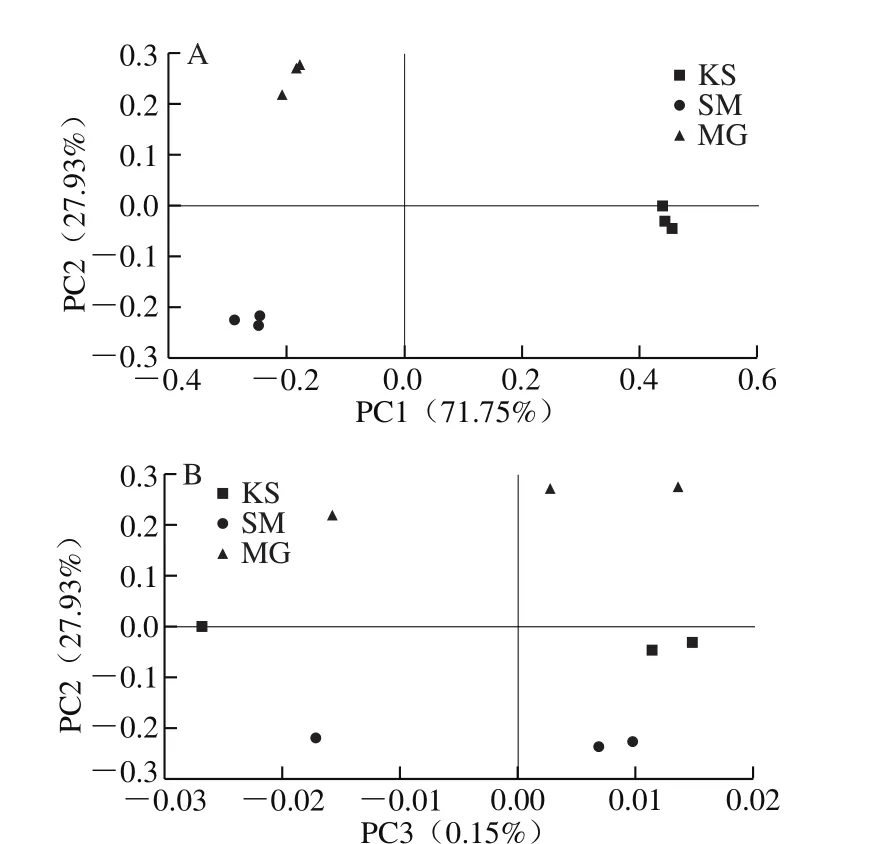
图5 曲拉中菌群结构UniFrac加权主坐标分析Fig. 5 Weighted UniFrac principal coordinate analysis of bacterial communities in Qula samples
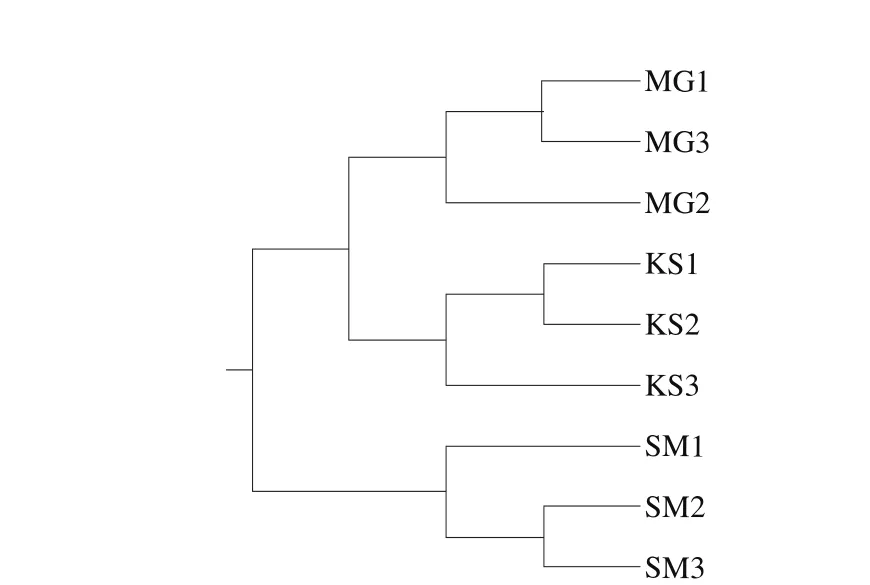
图6 基于Unweighted UniFrac距离矩阵的UPGMA聚类分析图Fig. 6 Dendrogram from UPGMA cluster analysis based on unweighted UniFrac distance matrix
2.8 样品中与细菌的基因代谢有关的功能特征

表4 与样品中细菌的基因代谢有关的功能特征Table 4 Functional features related to genes involved in metabolism from bacteria in Qula samples

表5 样本中细菌的其他功能基因Table 5 Other functional genes from bacteria in Qula samples
PICRUSt软件用于预测样品中存在的细菌类群的功能基因及其代谢途径[30]。对曲拉样本中的功能基因及代谢途径预测情况如表4所示。在样本中的功能基因中,44.04%~50.37%的微生物基因与以下代谢途径有关:氨基酸代谢、碳水化合物代谢、能量代谢、核苷酸代谢、辅助因子及维生素代谢、外源物质的生物降解与代谢、脂质代谢、酶家族、糖生物合成与代谢、其他氨基酸的代谢、萜类化合物与多酮类化合物的代谢以及其他次生代谢产物的生物合成。其中,碳水化合物代谢和氨基酸代谢的基因在曲拉样品中占主导地位,分别占所有基因的10.40%和8.68%。
样品中存在的细菌的其他功能基因与细胞处理、环境信息处理、遗传信息处理、人类疾病、机体系统等有关,具体情况如表5所示。其中,涉及膜转运的基因占主导地位,占所有基因的12.85%。
以不同样品中前50 种功能基因绘制热图,结果如图7所示。相同来源的曲拉样品之间的功能基因相似程度较高,而不同来源的样品的差异性较大,这一结果与前面细菌群落的聚类分析结果一致。

图7 基于样本前50 个功能基因的聚类分析热图Fig. 7 Heatmap from clustering analysis based on top 50 functional genes in Qula samples
3 结 论
本研究基于Illumina MiSeq高通量测序平台分析曲拉样品中细菌群落结构及多样性。结果表明,不同来源的曲拉样品中的微生物多样性存在差异性。与传统方法相比较,高通量测序的研究结果能够更全面地反映曲拉中菌群分布多样性。曲拉中细菌群落组成分析表明,曲拉样品中优势门为厚壁菌门和变形菌门;优势属为乳杆菌属、醋酸杆菌属、乳球菌属,细菌群落组成表明在不同来源的样品中群落组成存在差异。样品中存在的细菌的功能基因预测表明,不同来源样品的细菌群落之间存在差异。
