HPLC法测定银菊感冒胶囊中马来酸氯苯那敏的含量及含量均匀度
2019-12-03
云南省曲靖市食品药品检验检测中心,云南 曲靖 655000
银菊感冒胶囊为曲靖市中医医院的院内制剂(批准文号:滇ZJGF/2005-538),由金银花、菊花、射干、氨基比林、马来酸氯苯那敏等组成,具有清热解毒、解热镇痛的作用。临床用于感冒、发热、头痛、咽喉肿痛等症[1]。制剂中马来酸氯苯那敏仅有薄层色谱定性鉴别,未建含量测定方法。本文参照相关文献[2-4],采用高效液相色谱法(HPLC)对该品种中马来酸氯苯那敏含量及含量均匀度进行测定,结果表明本方法简单快捷,稳定性及重现性较好,可有效控制银菊感冒胶囊质量。
1 仪器与材料
1.1 仪器 Agilent1200型高效液相色谱仪(四元泵,自动进样器,柱温箱, 紫外检测器);METTLE MS205DU电子天平(上海梅特勒-托利多仪器有限公司)MS205DU电子天平(上海梅特勒-托利多仪器有限公司);一体式超纯水仪(PURELAB Flex 3,英国ELGA公司)。
1.2 药品与试剂 银菊感冒胶囊(规格0.25g/粒,批号181007、181118、181220)样品均来自云南省曲靖市中医医院;马来酸氯苯那敏(批号100047-201507,含量99.7%)购于中国食品药品检定研究院;乙腈为色谱纯,磷酸二氢铵和磷酸为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件与系统适用性试验 色谱柱:Amethyst C18(4.6 mm×200 mm,5 μm); 磷酸盐缓冲液(取磷酸二氢铵11.5 g,加水适量使溶解,加磷酸1 mL,用水稀释至1000 mL)-乙腈(80∶20)为流动相;流速:1.0 mL· min-1; 柱温:30 ℃,;检测波长:262 nm;进样量:10 μL。理论板数按氯苯那敏峰计算不低于4000,氯苯那敏峰与相邻杂质峰的分离度应符合要求。
2.2 溶液的制备
2.2.1 对照品溶液制备 取马来酸氯苯那敏对照品17.86 mg,精密称定,置200 mL容量瓶中,加流动相使溶解并稀释至刻度,摇匀,即得。
2.2.2 供试品溶液制备 取银菊感冒胶囊20粒,计算平均装量,取内容物,研细,混匀,取适量(约相当于马来酸氯苯那敏4 mg),精密称定,置50 mL容量瓶中,加流动性30 mL,超声处理15 min,放冷,加流动相至刻度,摇匀,滤过,滤液再用0.45 μm微孔滤膜滤过,即得。
2.2.3 阴性样品溶液制备 根据制剂处方及工艺,配制缺马来酸氯苯那敏的阴性样品(由云南省曲靖中医医院提供),再按“2.2.2”项下方法制备,即得。
2.3 结果
2.3.1 专属性试验 在上述色谱条件下,分别精密吸取对照品溶液、供试品溶液和阴性样品溶液各10 μL,注入液相色谱仪,记录色谱图,结果如图1所示。结果表明,马来酸氯苯那敏分离较好,无干扰,说明测定方法专属性较好。

2.3.2 线性关系试验 精密吸取“2.2.1”项下的对照品溶液0.1、0.2、0.5、1.0、2.0、5.0 mL,分别置于 10 mL容量瓶中,用流动相稀释至刻度,摇匀,即得。分别重复进样3次,记录峰面积。以进样量X(μg)为横坐标,峰面积(以氯苯那敏计)平均值(Y)为纵坐标,进行线性回归,得回归方程:Y=9226.8829X-0.6386,r=0.999 9。结果表明,马来酸氯苯那敏质量浓度在0.893~44.65 μg/mL范围内线性关系良好。
2.3.3 检测限和定量限 取“2.2.1”项下的对照品溶液适量,等倍逐级稀释进样测定,记录峰面积。当信噪比为3∶1时,得检测限为 2.025 ng;当信噪比为10∶1时,得定量限为 6.211 ng。
2.3.4 稳定性试验 取经“2.2.2”项方法制备的样品(批号181007)供试品溶液,按照“2.1”项下的色谱条件进行分析,分别在0、2、4、6、8、12、24 h不同时间进样测定。结果峰面积RSD为1.36%(n=7)。结果表明供试品溶液在 24 h 内色谱峰面积变化不大,溶液较稳定。
2.3.5 回收率试验 取已知含量的同一批样品(批号:181007)5份,每份约 0.5 g,精密称定;另取马来酸氯苯那敏对照品 25.12 mg,精密称定,置 25 mL容量瓶中,加流动性使溶解并稀释至刻度,摇匀,分别精密量取 2 mL置已称定样品中,按“2.2.2”项下的方法制备供试品溶液,按照“2.1”项下的色谱条件进行分析,结果见表1。
2.3.6 精密度试验 精密取经“2.2.1”项方法制备的样品(批号181007)供试品溶液 10 μL,按照“2.1”项下的色谱条件进行分析,连续6次。结果马来酸氯苯那敏峰面积的RSD分为0.30%,结果表明仪器平行精密度良好。
2.3.7 重复性试验 取同一批次的样品(批号:181007),平行6份(1.0 g),精密称定,按“2.2.1”项下的方法操作,按照“2.1”项下的色谱条件进行分析,每1 g样品中含马来酸氯苯那敏 3.9424 mg,RSD为0.26%,结果表明该方法重复性良好。
2.3.8 样品测定 含量测定:取样品3批次(批号181007、181118、181220)分别按“2.2.1”项下的方法制备供试品溶液,按照“2.1”项下的色谱条件进行分析,计算标示含量,结果3批样品中马来酸氯苯那敏的平均标示含量分别为98.56%、98.96%、99.05%。含量均匀度测定:取3个批号的样品各10粒,分别置于 25 mL容量瓶中,加流动性 15 mL,超声处理 15 min,放冷,加流动相至刻度,摇匀,滤过,续滤液再用 0.45 μm微孔滤膜滤过,滤液按照“2.1”项下的色谱条件进行分析,计算标示含量,结果见表2,符合《中国药典》2015年版通则0941含量均匀度项下有关规定(A+2.2S≤ L,L=15.0)。
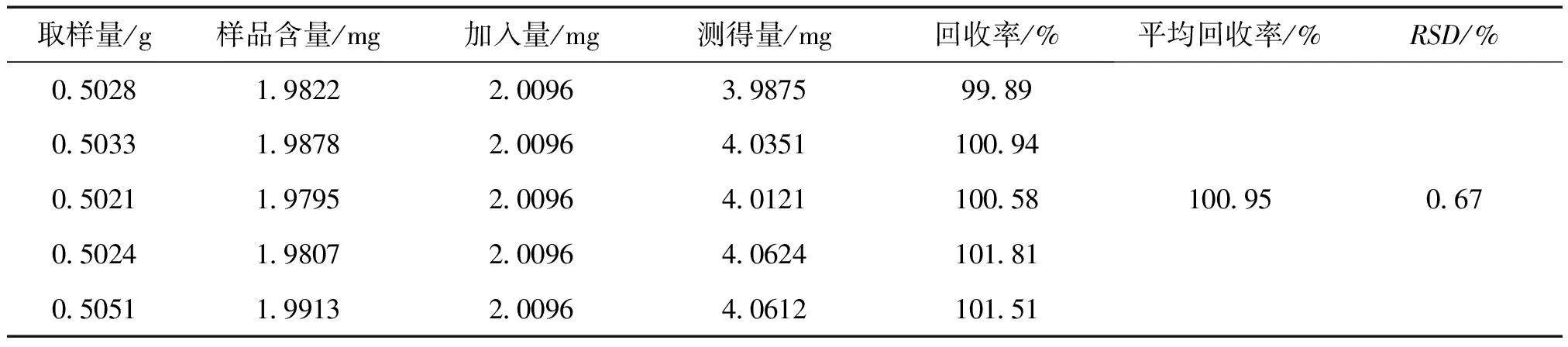
表1 回收率试验结果 ( n=5)

表2 样品含量均匀度测定结果 (n=3)
3 讨论
3.1 检测波长的选择 通过实验马来酸氯苯那敏在262.5 nm波长处有最大吸收,结合中国药典及相关文献报道[5-8]最终选用 262 nm作为检测波长。
3.2 提取条件选择 试验中曾采用甲醇、水、乙醇、流动相等溶媒超声15 min,进行提取,经比较,以流动相作为提取溶剂为佳;再以流动相为提取溶剂,对同一批号样品的超声时间进行考察,选择超声10、15、30、45、60 min后处理的样品溶液分别进行分析,结果各样品含量基本接近,故选择超声时间 15 min处理样品。
3.3 分离条件选择 本试验曾采用了3种流动相进行成分分离[9-11]。①乙腈:磷酸盐缓冲液②甲醇-水 ;③甲醇-乙腈-水。结果发现磷酸盐缓冲液(取磷酸二氢铵11.5 g,加水适量使溶解,加磷酸 1 mL,用水稀释至1000 mL)-乙腈(80∶20)为流动相保留时间合适且各成分获得较好的分离。
4 结论
本试验中采用高效液相色谱法测定银菊感冒胶囊中马来酸氯苯那敏的含量及含量均匀度,该方法操作简便,准确可靠,可有效控制该制剂质量。
