蛋白激酶C调节半乳凝集素-3促进心肌纤维化
2019-12-02王文超宋紫光
王文超 钟 晓 宋紫光 宋 湘
心力衰竭(简称心衰)是指由各种原因引起的心脏结构或功能改变,导致心室充盈和(或)射血功能受损的一组临床综合征[1]。心肌纤维化是由多种原因导致的正常心肌组织中细胞外基质沉积,胶原排列紊乱和成分比例失调为特点的疾病。心肌纤维化与多种心血管疾病密切相关,研究表明心力衰竭是心肌纤维化发生、发展的最终结果[2]。PKC为丝氨酸/苏氨酸激酶家族的成员,属于AGC激酶[3]。PKC的结构由NH2-末端、三磷酸腺苷和底物COOH-末端组成[4]。 PKC各亚型广泛分布于心肌细胞中,大量实验表明PKC在调控心肌细胞增殖、心肌肥厚、心肌保护和信号转导途径等过程中起着重要作用[5]。PKC在多种生理活动中都具有调节作用。当发生心力衰竭时,PKC表达明显升高[6]。Gal-3是由巨噬细胞激活分泌的一种蛋白,由单一基因LGALS3编码,位于14号染色体,它由一个非典型N端和一个C端碳水化合物识别域组成[7]。Gal-3在心肌巨噬细胞、肥大细胞浸润的过程发挥重要作用,能够促进心肌纤维化及心脏胶原蛋白沉积,促使心肌肥厚、心肌顺应性降低,最终导致心力衰竭的发生[8]。近年来研究表明,Gal-3是加重心力衰竭的一个重要因素,它参与心力衰竭的多种病理生理过程[9],但Gal-3是否参与PKC介导的心力衰竭目前报道甚少,本研究拟通过质粒构建、细胞转染和药物刺激来研究PKC和Gal-3是如何对心肌细胞胶原蛋白产生影响。
材料与方法
1.质粒构建:采用PCR扩增技术在大鼠肾脏cDNA中扩增大小789bp的Gal-3翻译区序列,并连接到p3xFLAG-CMV-10载体中。过程为Gal-3翻译区基因序列合成含HindⅢ和XbaⅠ位点的Gal-3上游及下游引物,Gal-3基因设计引物(Accessory number NM_031832),包含Hind Ⅲ位点的上游引物:5′-CAAGCTTATGGCAGACGGCTTCTCACTTAATG-3′;包含XbaⅠ位点的下游引物:5′-CTCTAGACTTAGATCATGGCGTGGGAAGCGCT-3′以pTA-Gal-3为模板PCR扩增Gal-3基因,经HindⅢ和XbaⅠ酶切后连接到p3xFLAG-CMV-10载体上。通过核苷酸序列分析鉴定构建的质粒。pcDNA3-PKC-α的构建如之前构建进行[10]。
2.心肌细胞培养:选择小鼠HL-1心肌细胞,该细胞株由美国路易斯安那州立大学William.Claycomb教授惠赠。将该细胞培养在含有4mmol/L L-谷氨酰胺、10%胎牛血清、0.1mmol/L去甲肾上腺素和1%青霉素的Claycomb培养基中,在饱和湿度、37℃、5%CO2条件下传代培养。在37℃、5%CO2培养箱中将培养瓶和接种板涂上纤维连接蛋白,每36h更换培养基,根据实验需要对其进行细胞转染和药物刺激。
3.细胞转染和药物刺激:将HL-1心肌细胞转移至6孔板,使心肌细胞生长至90%融合,选择Lipofectamine 2000按说明书方法转染质粒,48h后收集蛋白进行Western blot法检测。 需要添加PKC激活剂和PKC抑制剂进行处理细胞时,使HL-1心肌细胞生长至90%融合,饥饿处理12h后,加入PDB、白屈菜红碱(chelerythrine,Chele)。
4.Western blot法检测:收集心肌细胞,提取总蛋白,BioRad蛋白分析测得样品蛋白浓度。将相同质量的蛋白(50~100μg/lane)加于SDS-PAGE胶,经电泳分离蛋白后,经转膜,封闭,孵育相应一抗,PBST洗膜,孵育相应二抗,PBST洗膜后经ECL系统发光、显影。

结 果
1.激活PKC诱导Gal-3在心肌细胞的表达:分别用浓度为10-9、10-8、10-7和10-6mol/L的PKC激活剂PDB来刺激HL-1心肌细胞,24h后,收集蛋白,检测Gal-3的表达水平,24h后,Gal-3表达随着PDB浓度的增加而增加。选用PKC激活剂PDB和抑制剂Chele分别激活和抑制Gal-3,RIPA裂解细胞收集蛋白,Western blot法检测可见PDB组Gal-3表达显著增加54%(P=0.000),PKC抑制剂Chele降低Gal-3蛋白表达79%(P=0.000),详见图1。

图1 激活PKC诱导半乳凝集素-3在心肌细胞(HL-1)表达A.不同浓度的PDB刺激心肌细胞,收集蛋白,应用Western blot法检测半乳凝集素-3表达量;B.分别用二丁酸佛波醇酯、白屈菜红碱刺激心肌细胞,收集蛋白,应用Western blot法检测半乳凝集素-3表达量;与对照组比较,*P<0.05,**P=0.000,n=4
2.高表达Gal-3基因诱导胶原蛋白沉积:当6孔板的细胞生长至90%融合后,按说明书进行p3xFLAG-Gal-3质粒1微克/孔的转染,48h后进行Western blot法检测Gal-3的表达水平。高表达Gal-3基因诱导Ⅰ型胶原蛋白积累增加58%(P<0.01,n=3),详见图2。

图2 高表达Gal-3基因诱导胶原蛋白沉积HL-1细胞转染p3xFLAG-Gal-3,收集蛋白,Western blot法检测半乳凝集素-3、Ⅰ型胶原蛋白表达水平;与对照组比较,*P<0.05,**P<0.01,n=3
3.PKC诱导心肌细胞胶原沉积:待细胞长至90%融合,饥饿处理12h,按照Lipofectamine 2000说明书进行pcDNA3-PKC-α质粒的转染,48h后通过Western blot法检测PKC的表达水平(P<0.05)。如图3所示,表达PKC基因诱导Ⅰ型胶原蛋白积累增加54%(P<0.01)。
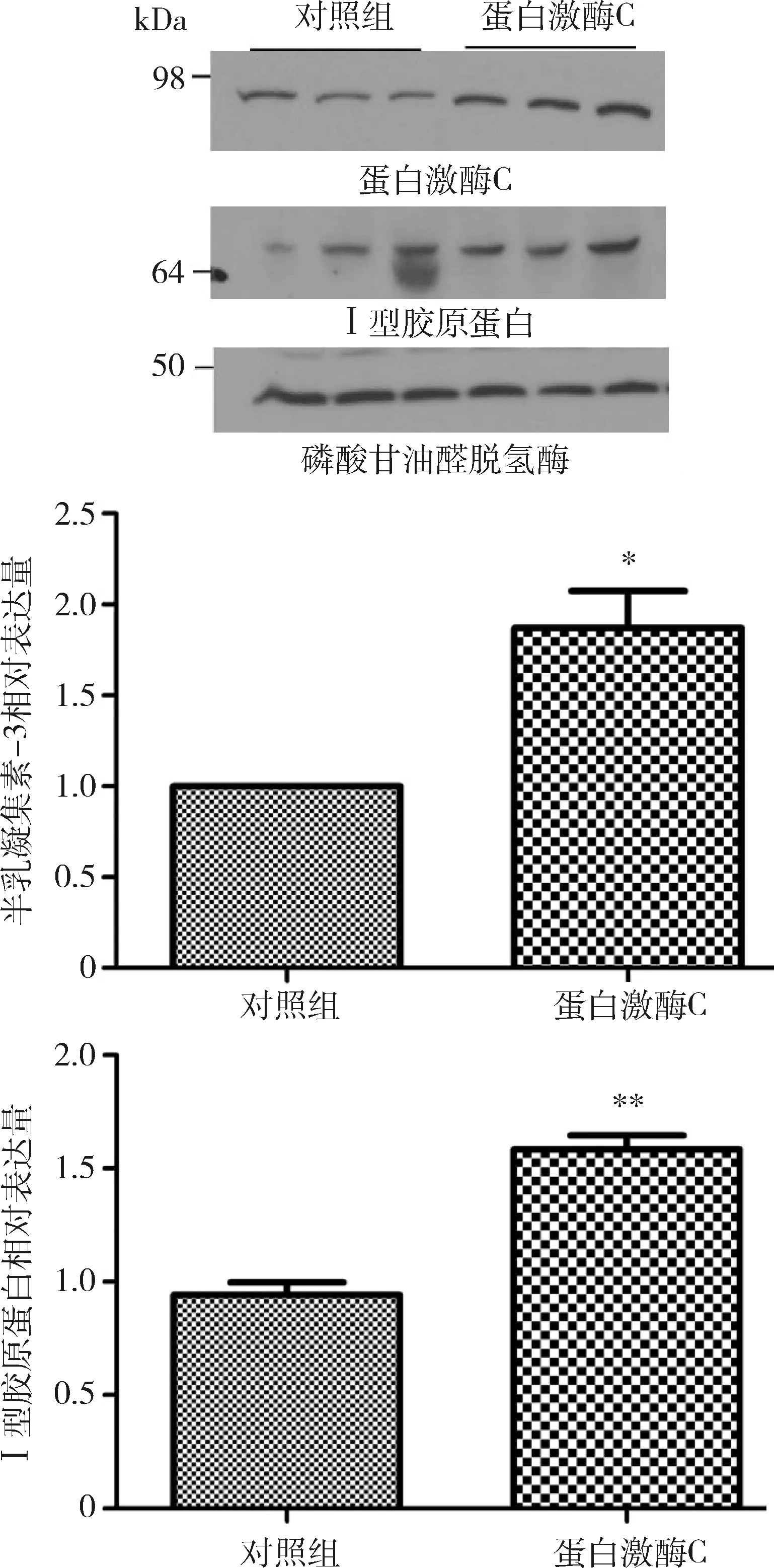
图3 PKC诱导心肌细胞胶原沉积HL-1细胞转染pcDNA3-PKC-α,Western blot法检测蛋白激酶C、Ⅰ型胶原蛋白的表达水平;与对照组比较,*P<0.05,**P<0.01,n=3
4.抑制Gal-3下调PKC诱导的心肌细胞胶原沉积:高表达的Gal-3可诱导心肌细胞胶原沉积,为进一步研究Gal-3是否介导PKC诱导胶原沉积的过程,应用Gal-3抑制剂β-乳糖50mmol/L和PKC激活剂PDB 2μmol/L处理心肌细胞6h;第4组先应用β-乳糖50mmol/L处理心肌细胞2h,再用PDB 2μmol/L处理心肌细胞。Western blot法检测可见抑制Gal-3下调PKC诱导的心肌细胞胶原沉积降低24%(P<0.05),详见图4。
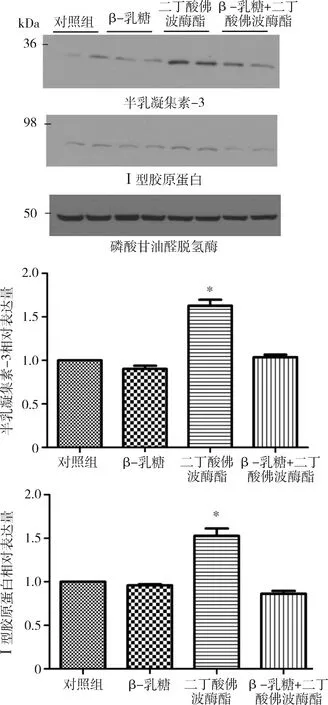
图4 抑制Gal-3下调PKC诱导的心肌细胞胶原沉积HL-1细胞分别应用β-乳糖、二丁酸佛波醇酯以及β-乳糖+二丁酸佛波醇酯处理心肌细胞,收集蛋白,Western-blot法检测半乳凝集素-3、Ⅰ型胶原蛋白的表达水平;与对照组比较,*P<0.05,*P=0.000,n=5
讨 论
PKC是由Nishizuka等于1977年首次发现的一组磷脂依赖性激酶。PKC分为3类12种同工酶,第1类为经典型PKC,第2类为新型PKC,第3类为不典型PKC,不同亚型在机体组织的分布也不同。心肌细胞表达多种PKC亚型,PKC参与调节生物体多种病理生理过程[11]。然而心肌肥厚时心肌细胞内表达增加的主要是PKC信号蛋白,这说明PKC在心脏病理改变时发挥重要作用。心力衰竭动物模型实验发现PKC过表达可引起心肌肥大和心功能障碍,由此证明PKC 参与了心肌纤维化过程[12]。PKC在心肌纤维化和心力衰竭发病过程中的促进作用已经被证实,然而其参与的信号通路和发病机制目前尚未清楚。研究发现,一些特殊物质如丝裂原活化蛋白激酶(MAPK)、细胞外调节蛋白激酶 (ERK)、糖原合成酶激酶(GSK)均为PKC的靶向作用物质,并且这些物质均参与心肌纤维化过程[13~15]。
Gal-3是动物凝集素家族中的一员,属于钙的非依赖性糖结合蛋白,是人类基因组中唯一的嵌合型半乳糖凝集素[16]。心肌重构是心室结构和功能变化的病理过程,是心力衰竭的前期阶段,是病变发展和预后不良的决定性因素。心肌纤维化是心肌重构发生、发展的关键,然而,Gal-3 是心肌纤维化的关键介质,通过介导心肌细胞外基质逐渐促进心力衰竭的发生和发展。最近实验表明Gal-3通过多种途径诱发心力衰竭,例如通过转化生长因子β介导心肌胶原纤维不对称的沉积[17];诱导纤维母细胞转化为肌成纤维细胞,增加心肌细胞外胶原的沉积;降低细胞内的抗氧化能力,增加细胞的凋亡,这些机制促进心肌纤维化,心肌重构,最终发展为心力衰竭。Gal-3参与心脏、肝脏等各器官纤维化的发生和发展,并最终发展为器官衰竭[18,19]。
以上实验证明了PKC和Gal-3在心力衰竭心肌纤维化的过程中起了关键性作用。两者都是通过促进胶原蛋白的沉积来促进心力衰竭的发展,但两者在心力衰竭发展过程中的关系尚未清楚,笔者通过体外实验发现,PKC对Gal-3的表达具有促进作用,并且Gal-3的表达与PKC的浓度呈正相关,但两者之间具体调节机制尚不清楚,本实验通过抑制Gal-3的表达发现PKC对胶原蛋白沉积的诱导作用减弱。
许多实验已经证实Gal-3激活或表达增加与心力衰竭密切相关,实验发现PKC对Gal-3的调节在心肌纤维化和心力衰竭发展过程中起着关键作用[20]。因此提出增加PKC可以促进Gal-3的表达,进而促进心肌纤维化和心力衰竭的发展。本实验是在现有研究基础之上的进一步拓展,提出存在PKC-Gal-3通路,且该通路在心力衰竭的发生、发展中起着关键作用。而此通路可能为预防、延缓或治疗心力衰竭提供新的理论依据,然而对于此通路的其他调节尚不明确,因此需要进一步探究是否还有其他物质直接参与该通路的调节。
