磷钨钒杂多酸相转移催化剂的制备及其深度氧化脱硫性能
2019-11-22鄢景森王泽青鄂永胜唐俊杰艾丽梅高军峰
鄢景森, 王泽青, 鄂永胜, 唐俊杰, 艾丽梅, 高军峰
(辽宁科技学院 药化学院, 辽宁 本溪 117004)
柴油中的含硫化合物燃烧后,所形成的硫氧化合物(SOx)是引发酸雨、雾霾的重要原因。当前,石油炼制行业应用最多的燃油脱硫方法为催化加氢,但加氢脱硫工艺虽然能够脱除柴油中的硫醇、硫醚、噻吩及其衍生物,但对位阻效应较大的二苯并噻吩及其衍生物脱除率较低。为了得到符合中国国V标准的超低硫燃料油(<10 μg/g),必需在更低的空速、更高的温度和压力等条件下进行,大大增加了生产成本,因此,研究者一直在寻求其他非加氢深度脱硫的方法,如氧化脱硫、吸附脱硫、萃取脱硫、烷基化脱硫和生物脱硫等[1,2]。与传统的加氢脱硫技术相比,氧化脱硫技术具有投资少,反应条件温和,不需要昂贵的氢源,对加氢脱硫方法难以脱除的二苯并噻吩及其衍生物表现出很高的氧化脱硫活性,有利于实现深度脱硫等优点[2-4]。
催化氧化脱硫体系中选择H2O2作为氧化剂的研究较多,主要是因为H2O2的氧化产物为水,是清洁和环境友好的氧化剂,如甲酸/ H2O体系[5]、乙酸/ H2O催化体系[6]。分子筛/H2O2体系[7]、杂多酸(盐)/H2O2体系[8,9]和功能化离子液体/H2O2体系[10-13]。其中,杂多酸(盐)催化剂是一种兼具酸性、氧化还原性的双功能催化剂,除了具有独特的六方笼状结构和“假液相行为”的特点之外,催化性能还可以通过改变元素组成和结构来调变,从而可以满足不同的应用和需要[14-17]。但杂多酸(盐)/H2O2体系是在两相系统中进行的,水相中的H2O2与燃油中的含硫化合物接触面积小,相间阻力大,传质速率低,影响氧化反应速率和脱硫效果。季铵盐相转移催化剂的应用则可以帮助反应物从一相转移到能够发生反应的另一相当中,增大两相接触面积,减小反应阻力,降低反应温度,加快异相系统反应速率[18,19]。杂多酸与季铵盐结合成相转移催化剂一般有两种形式,一种是杂多酸与季铵盐以混合物形式进入反应体系,结合成离子对直接作为催化剂使用,如Huang等[20]采用这种方式考察了不同链长的双亲性季铵盐对脱硫效率的影响,发现加入较长碳链的鎓盐比较短碳链的鎓盐的催化活性更好,而且不同链长的鎓盐对催化剂分离回收难易程度也有影响。另外一种方式是杂多酸和季铵类表面活性剂反应,通过带正电荷的表面活性剂取代杂多酸酸抗衡离子制备成具有相转移功能的固体催化剂,如李灿课题组采用这种方式先后制备了双亲性催化剂[(C18H37)2N(CH3)2]3PW12O40和[C18H37N(CH3)3]PV2MoO40,使用不同的亲油性阳离子表面活性剂集团包裹不同的杂多阴离子基团形成微乳体系,催化剂与油相中的含硫化合物充分接触,大大缩短了反应时间,显著提高了杂多酸的催化效率[21-25]。杂多酸的氧化脱硫与中心原子或配位原子有关, Te等[16]研究发现,不同种类杂多酸对DBT类含硫化合物的氧化脱硫活性顺序为:磷钨酸>磷钼酸>硅钼酸>硅钨酸,磷钨酸表现出最高的活性。在氧化性方面,V比Mo和W有更高的氧化性[26]。目前,有关钒取代磷钨杂多酸相转移催化剂的报道较少,本研究尝试将磷、钨与钒混合配位得到氧化能力更高的磷钨钒三元杂多酸,然后利用不同烷基链的季胺盐表面活性剂与其进行离子交换,得到了一系列不同阳离子的相转移催化剂,在以H2O2为氧化剂的模拟油品的深度氧化脱硫反应中,考察了该催化剂的氧化脱硫活性、重复使用性能和可回收性能,探索了最优的催化剂组成和反应条件,取得了较好结果。
1 实验部分
1.1 仪器与试剂
偏钒酸钠、钨酸钠、磷酸氢二钠、硫酸、无水乙醇、无水乙醚、磷钨酸、四甲基溴化铵、四丁基溴化铵、四正辛基溴化铵、十四烷基三甲基溴化铵、十六烷基三甲基溴化铵、十八烷基三甲基溴化铵、二苯并噻吩、苯并噻吩、4,6-二甲基二苯并噻吩、N,N-二甲基甲酰胺、双氧水(30%),均为分析纯,购自国药集团化学试剂有限公司。
采用赛默飞ICAP-7200型电感耦合等离子体发射光谱仪进行元素分析。采用北京瑞利WQF-510A傅里叶变换红外光谱仪测定红外光谱,KBr压片,400-4000 cm-1扫描。采用Bruker D8 Advance 型X射线粉末衍射仪上测定X射线衍射光谱,扫描速度0.02(°)/s, 4°-40°扫描,射线源为CuKα(λ=0.154 nm),工作电压40 kV,电流40 mA。
1.2 催化剂的制备
1.2.1 11-钨-1-钒磷酸(H4[PW11VO40])的合成
H4[PW11VO40]参照文献[27]方法合成。将0.055 mol钨酸钠(Na2WO4·2H2O),0.005 mol磷酸氢二钠(Na2HPO4·12H2O)分别溶于热水中并混合,再与溶解于热水中的0.005 mol钒酸钠(NaVO3)混合,加入49%硫酸调节pH值至2.0-2.5,在90 ℃下搅拌反应2 h。 然后将热的反应液冷却至常温,转移至250 mL分液漏斗中,加入30 mL乙醚并振荡。然后缓慢逐滴加入49%硫酸,每加入2.0-3.0 mL就进行振荡,静置,至溶液分出三层,萃取出最下层黄色油状物,重复几次,合并萃取液。50 ℃下水浴加热挥去乙醚后将其放入50 ℃真空干燥箱中加热烘干,得到淡黄色固体产物。H4[PW11VO40]中主要元素P、W、V分析结果与理论含量(括弧内值表示)为:P,1.22%(1.13%);W,70.60%(73.60%);V,1.65%(1.85%),测定值与理论值基本吻合。
1.2.2 磷钨钒杂多酸相转移催化剂的合成
阳离子表面活性剂与磷钨钒杂多酸按不同的物质的量比反应,可得到不同组成的磷钨钒杂多酸相转移催化剂。以十六烷基三甲基溴化铵(CTAB)与H4[PW11VO40]为例说明,分别将0.8、0.6、0.4、0.2 mmol CTAB溶于20 mL蒸馏水中形成溶液A, 将0.55 g(0.2 mmol) H4[PW11VO40]溶于30 mL蒸馏水中形成溶液B, 将溶液B逐滴加入溶液A中产生乳白色沉淀物,在50 ℃下搅拌5 h。在室温下冷却后,抽滤,用去离子水洗涤沉淀至无Br-为止,所得粉末固体在105 ℃烘箱中干燥即可得到不同组成的催化剂,分别记为(CTAB)4[PW11VO40]、(CTAB)2H2[PW11VO40]、(CTAB)3H[PW11VO40]和(CTAB)H3[PW11VO40]。
将磷钨钒杂多酸与不同种类的阳离子表面活性剂按1∶3物质的量比投料反应,可得到一系列不同种类的磷钨钒杂多酸相转移催化剂,阳离子表面活性剂包括四甲基溴化铵(TMAB)、四丁基溴化铵(TBAB)、四正辛基溴化铵(TOAB)、十四烷基三甲基溴化铵(TTAB)和十八烷基三甲基溴化铵(STAB),制备方法与上面类似,所得催化剂分别记做(TMAB)3H[PW11VO40]、(TBAB)3H[PW11VO40]、(TOAB)3H[PW11VO40]、(TTAB)3H[PW11VO40]和(STAB)3H[PW11VO40]。
1.3 催化剂活性的评价
氧化脱硫反应在带有磁力搅拌装置和回流冷凝管的三口烧瓶中进行。以一个典型的实验说明:向三口烧瓶中加入40 mL硫质量分数为1000 μg/g 二苯并噻吩(DBT)的正辛烷溶液(模拟油原料),加入一定量的催化剂和过氧化氢(H2O2), 磁力搅拌,在恒温水浴50 ℃下反应1.0-3.5 h。每隔0.5 h吸取少量反应液一次,离心分离催化剂固体后,量取少量上层清液进行测定,采用Agilent 6890N气相色谱仪分析含硫化合物含量, HP-5毛细管分析柱( 30 m×0.32 mm×1.0 μm),氢火焰离子化(FID)检测器,分流进样,程序升温,初始100 ℃,保持3 min,然后以10 ℃/min升至260 ℃,保持3 min。外标法定量,催化剂性能以DBT的转化率(x)表示:
(1)
式中,C0为反应初始DBT含量,CR为反应后体系中DBT含量。
3)酒店部分7~9层由地下1层酒店生活水池及裙房变频给水设备联合供水;地下1层设酒店客房工频加压泵,抽水至25层(避难层)酒店客房低区生活水箱,该水箱另设一组工频加压泵抽水至屋顶层酒店客房高区生活水箱。酒店客房给水分成2个区,11~17层为低区,由25层酒店客房低区生活水箱重力供水;18~24层为高区,由屋顶层酒店客房高区生活水箱重力供水。
2 结果与讨论
2.1 红外(FT-IR)表征
图1为H4[PW11VO40]和代表性相转移催化剂(CTAB)3H[PW11VO40]反应前后的红外吸收光谱谱图。由图1可知,H4[PW11VO40]在804、893、982、1080 cm-1处出现了Keggin结构杂多酸的四个特征吸收峰,分别归属为M-Oc-M键的伸缩振动、M-Ob-M的伸缩振动、M=Od键的伸缩振动、P-Oa键的伸缩振动,其中,Oa是与中心原子P相连的氧, Ob为不同三金属簇间连接的桥氧,Oc为相同三金属簇内连接的桥氧,Od为与配位原子(W或V)相连的端基氧[27-29]。1616和3402 cm-1处的特征吸收峰,则分别归属于O-H键的变形振动和伸缩振动,这主要来源于杂多酸表面吸附的结晶水[27]。CTAB的红外光谱谱图中,2920和2849 cm-1处的吸收峰分别归属为-CH2和-CH3的伸缩振动,1475 cm-1为-CH变形振动峰。新制备的(CTAB)3H[PW11VO40]也出现了杂多酸Keggin结构的特征衍射峰,以及2920、2849、1475 cm-1的特征吸收峰,由此可以确定相转移催化剂中烷基和杂多阴离子基团的存在,说明CTAB和磷钨钒杂多酸成功完成了离子交换,并分别保留了原来各自基团的结构特征。与新制催化剂相比,反应后催化剂的红外光谱谱图几乎没有变化,说明催化剂具有良好的稳定性。
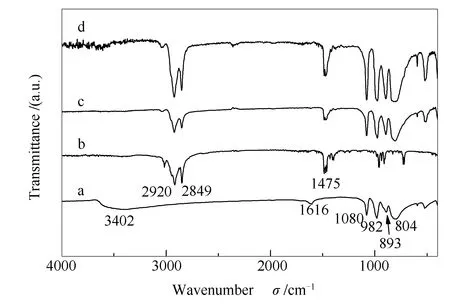
图 1 样品的红外光谱谱图Figure 1 FT-IR spectra of the samples a: H4[PW11VO40]; b: CTAB; c: fresh (CTAB)3H[PW11VO40]; d: spent (CTAB)3H[PW11VO40]
2.2 X射线衍射(XRD)表征
图2为H4[PW11VO40]和(CTAB)3H[PW11VO40]的XRD谱图,由图2可知,两者的XRD谱图有着明显区别,H4[PW11VO40]在7°-11°、18°-22°、24°-30°、32°-38°出现了Keggin结构杂多酸晶体的特征衍射峰[27-29]。杂多酸季胺盐(CTAB)3H[PW11VO40]除了在低角衍射区有明显的衍射峰存在,其杂多阴离子特征峰的强度在广角衍射区明显减弱至几乎看不到衍射峰,这说明磷钨钒杂多酸的氢离子被CTAB阳离子取代后杂多酸阴离子的一级结构没有变化,但杂多化合物的二级结构有了明显变化,季铵盐阳离子的引入促进了复合材料中阴离子簇的分散,这一现象是在杂多阴离子和有机阳离子之间的静电相互作用及范德华力共同作用下形成的[30]。
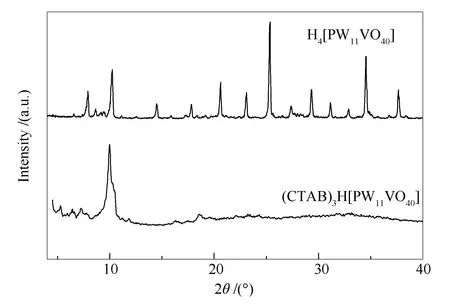
图 2 H4[PW11VO40]和(CTAB)3H[PW11VO40]的XRD谱图
Figure 2 XRD patterns of H4[PW11VO40] and (CTAB)3H[PW11VO40]
2.3 催化剂性能评价与分析
2.3.1 不同催化剂的氧化脱硫性能评价
评价了磷钨、磷钨钒杂多酸及其相应的杂多酸相转移催化剂对DBT的氧化脱硫性能,结果见表1。由表1可知,不加催化剂时(编号1),DBT转化率为3.37%,这说明没有催化剂存在时反应速率很慢。在未引入季铵盐表面活性剂阳离子时,H3[PW12O40]和H4[PW11VO40]催化剂(编号2、3)的DBT转化率分别为24.38%和36.15%,转化率不高的原因主要归因于杂多酸/H2O2体系中,水油两相传质阻力大,影响了反应速率和脱硫效率。杂多阴离子的不同对催化活性有一定影响。H4[PW11VO40]催化剂活性要高于H3[PW12O40]催化剂,类似地,钒取代的杂多酸相转移催化剂(CTAB)3[PW11VO40]催化活性也要高于(CTAB)3[PW12O40]的反应活性,这一结果可以归因于配位原子V比W原子具有更高的氧化性,这一结果与Yamaura等[31]报道的相一致。在相同条件下,以(CTAB)3[PW12O40]和(CTAB)3H[PW11VO40]为催化剂时,DBT脱除率分别是H3[PW12O40]和H4[PW11VO40]催化剂的2.96倍和2.39倍(编号2-5),这说明季铵盐相转移杂多酸催化剂降低了两相传质阻力,增大了相界面接触面积,显著地提高脱硫效率。
由表1可以看出,不同碳链长度的季铵阳离子对磷钨钒杂多酸相转移催化剂的催化活性有显著影响,各相转移杂多酸催化剂的脱硫性能大小依次为:(CTAB)3H[PW11VO40]≈(STAB)3H[PW11VO40]>(TTAB)3H[PW11VO40]>(TOAB)3H[PW11VO40]>(TBAB)3H[PW11VO40]>(TMAB)3H[PW11VO40],说明杂多酸相转移催化剂的活性与季胺阳离子种类密切相关,(TMAB)3H[PW11VO40]的脱硫性能最差,这是因为四甲基溴化铵(TMAB)的亲酯性不强,而碳链较长的十六烷基三甲基溴化铵(CTAB)或十八烷基三甲基溴化铵(STAB)季胺阳离子的亲脂性较强,有利于包裹DBT分子,形成稳定均一的乳液滴,并使之与催化活性中心有充分的接触,从而有更高的反应活性[32]。(CTAB)3H[PW11VO40]催化剂的活性与(STAB)3H[PW11VO40]相当,但CTAB的价格还不到STAB的二分之一,具有更好的经济优势,故本实验筛选确定的最优催化剂为CTAB与H4[PW11VO40]离子交换合成的催化剂(CTAB)3H[PW11VO40],并选择该催化剂去考察反应条件对DBT转化率的影响。
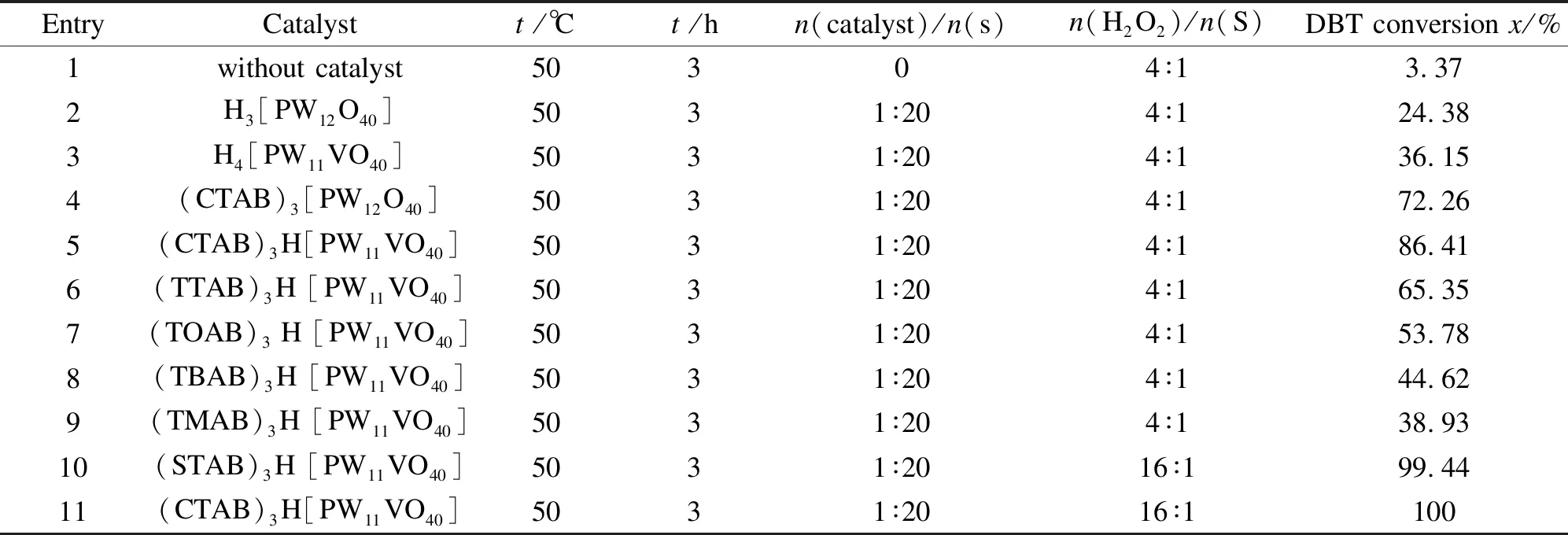
表 1 不同催化剂对DBT氧化脱硫性能的比较
相转移催化氧化脱硫反应的机理可以用图3来解释。杂多酸阴离子Y-与季铵盐阳离子Q+以离子对的形式存在于反应体系中,杂多酸阴离子Y-是亲水基团,被水相中的H2O2氧化变成活性的过氧杂多酸阴离子Y-[O],季铵盐阳离子Q+是亲油基团,可将氧化后的杂多酸阴离子Y-[O]带入油相中,催化氧化油中的含硫化合物DBT,使之变成极性较大的二苯并噻吩砜,而过氧杂多酸阴离子则还原为杂多阴离子,重新进入水相中,完成一个催化循环。
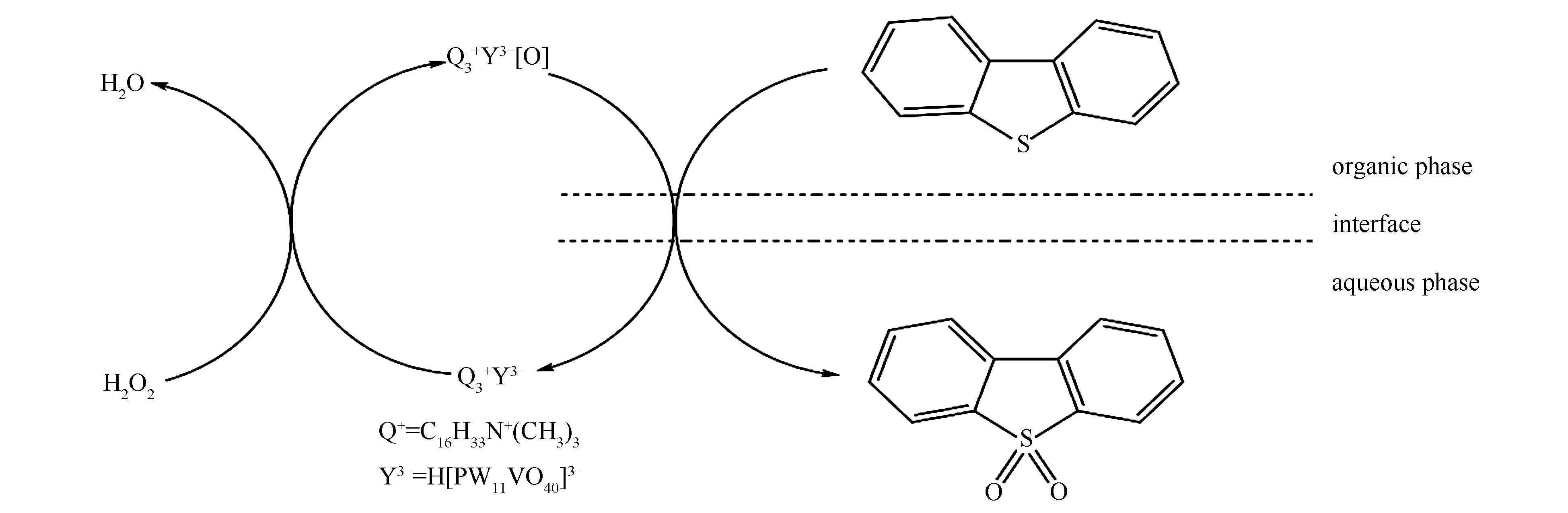
图 3 (CTAB)3H[PW11VO40]对DBT的氧化脱硫反应机理示意图
2.3.2 催化剂不同组成对DBT转化率的影响
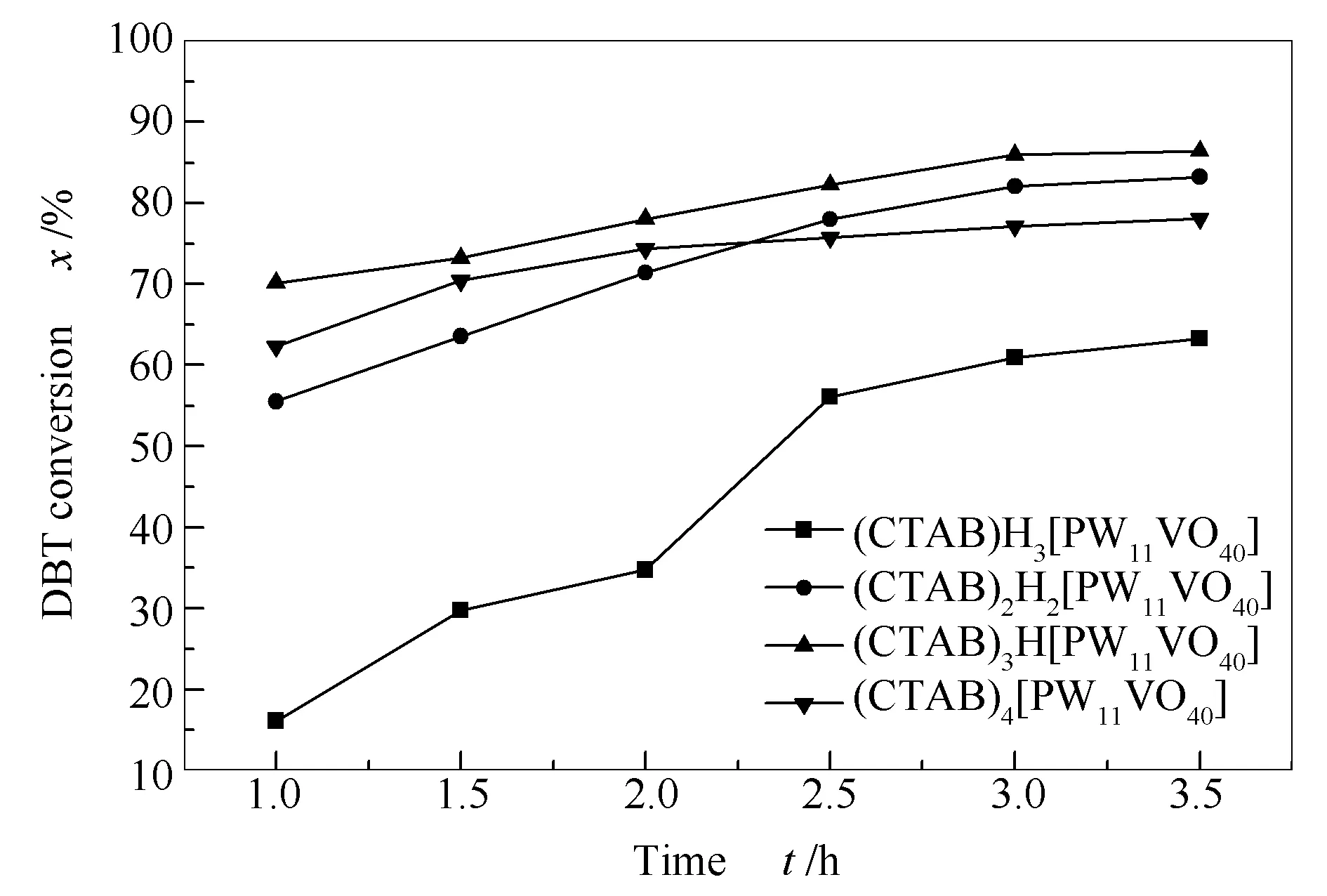
图 4 不同催化剂组成对DBT转化率的影响Figure 4 Effect of catalyst composition on the conversion of DBT
2.3.3 催化剂不同用量对DBT转化率的影响
(CTAB)3[PW11VO40]催化剂用量按n(catalyst)/n(S)的物质的量比计算,其不同物质的量比对DBT转化率的影响见图5。由图5可知,1 h内各催化剂的用量越小,其起始DBT转化率越低,随着反应时间延长,除物质的量比为1∶100的催化剂之外,其余催化剂在3 h后都可以实现99%以上的脱硫,所以综合DBT转化率和节省催化剂用量角度出发,选择物质的量比为1∶80的催化剂为最佳投料用量。
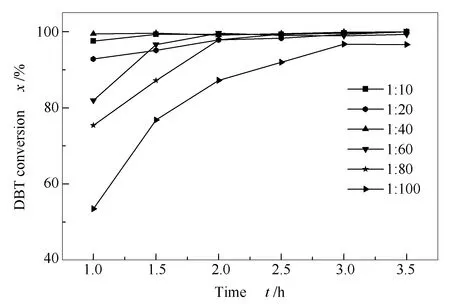
图 5 催化剂不同用量n(catalyst)/n(S) 对DBT转化率的影响
2.3.4 不同氧硫比对DBT转化率的影响
图6为不同氧硫比(n(H2O2)/n(S))对DBT转化率的影响,由图6可知,氧硫比为2时,3 h时DBT转化率为82.67%;氧硫比为4∶1时,DBT转化率有所增加,但不显著;氧硫比为8∶1时,脱硫率显著增加,1 h时转化率为85.32%,3 h时的脱硫率可以达到100%;继续增加氧硫比至16∶1和32∶1时,反应速率加快, DBT的转化率经1 h即可达到94.85%和98.66%,但3 h后与氧硫比为8∶1的转化率相当。 理论上,完全氧化1 mol的DBT需要2 mol的H2O2,但由于H2O2在反应时往往存在无效的热分解反应,此外,很可能H2O2在氧化该催化剂生成真正起催化作用的过氧杂多阴离子时,需要更多量的活性氧[33],另一方面,所选用的催化剂(CTAB)3H[PW11VO40]不溶于水相,因此,较高的氧硫比可以提高H2O2浓度,提高扩散和传质速率,有利于杂多酸阴离子氧化生成更多的过氧杂多阴离子活性物种,故本实验氧硫比8∶1为适宜的氧硫比。
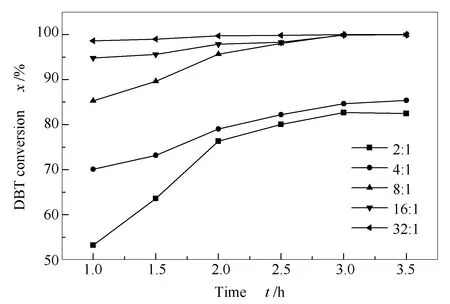
图 6 不同n(H2O2)/n(S)物质的量比 对DBT转化率的影响
2.3.5 反应温度和反应时间对DBT转化率的影响
考察了在不同反应温度、反应时间时催化剂(CTAB)3H[PW11VO40]对DBT的转化率,结果见图7。由图7可知,DBT的转化率随着反应温度增高而增大,但并不是线性增加。对比2.5 h内的DBT转化率可以发现,30 ℃反应时,DBT的转化率为88.86%;当反应温度分别增加至40和50 ℃时,DBT转化率分别为95.76%和99.46%;当反应温度由50 ℃增加至60 ℃时,催化剂的转化率为99.68%,仅仅增加了0.22%。由图7还可知,DBT转化率随反应时间延长而增大,但对于不同的反应温度,催化剂达到化学平衡所用的时间不同,催化剂在3 h内DBT的转化率增加较快,3 h后DBT转化率已基本不变,说明反应3 h后已趋近于达到热力学平衡。上述分析说明,在一定范围内,提高反应温度、延长分析时间有助于DBT的氧化脱硫反应,所以从反应平衡时间和减少能耗出发,选择50 ℃和3 h为最佳的反应温度和反应时间。
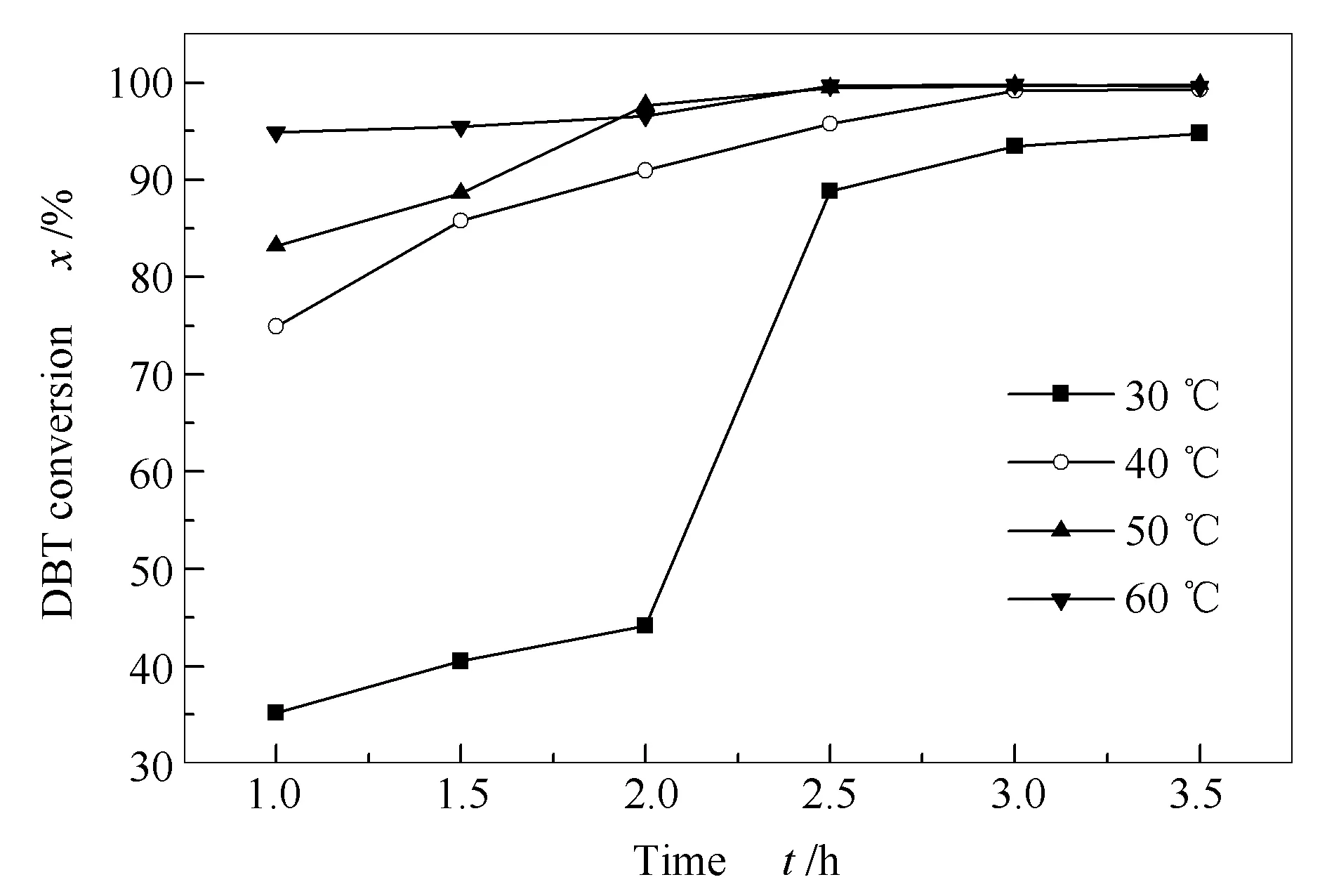
图 7 不同反应温度对DBT转化率的影响
2.3.6 催化剂重复使用性能的考察
(CTAB)3H[PW11VO40]催化剂对DBT氧化脱硫反应过程见图8,由图8可知,在刚刚反应时催化剂为固态,且不溶于反应底物,沉在反应器底部(图8(a));当温度升至反应温度50 ℃时,在搅拌情况下,反应体系变成乳状液,成为单一的均相(图8(b));反应结束后冷却静置,催化剂恢复至固态沉在反应器底部,通过过滤方法就可轻易使反应产物和催化剂得以分离,方便回收(见图8(c))。测定催化剂重复使用性能时,是采用倾析的方法小心将脱硫后的反应液倾出,催化剂留在反应器底部,再向三颈瓶内加入同样的反应物进行反应。图9是催化剂在最优条件下重复使用五次的反应结果。可以看出,随着反应次数增加,DBT转化率几乎未有变化,说明该催化剂具有良好的重复使用性能。在分离后的液体产物中加入极性溶剂N,N-二甲基甲酰胺(DMF),则油相和DMF快速明显分层,反应后油相中生成的砜类被萃取到DMF中,DMF可通过蒸馏的方法反复利用。

图 8 (CTAB)3H[PW11VO40]催化剂对DBT氧化脱硫反应过程图
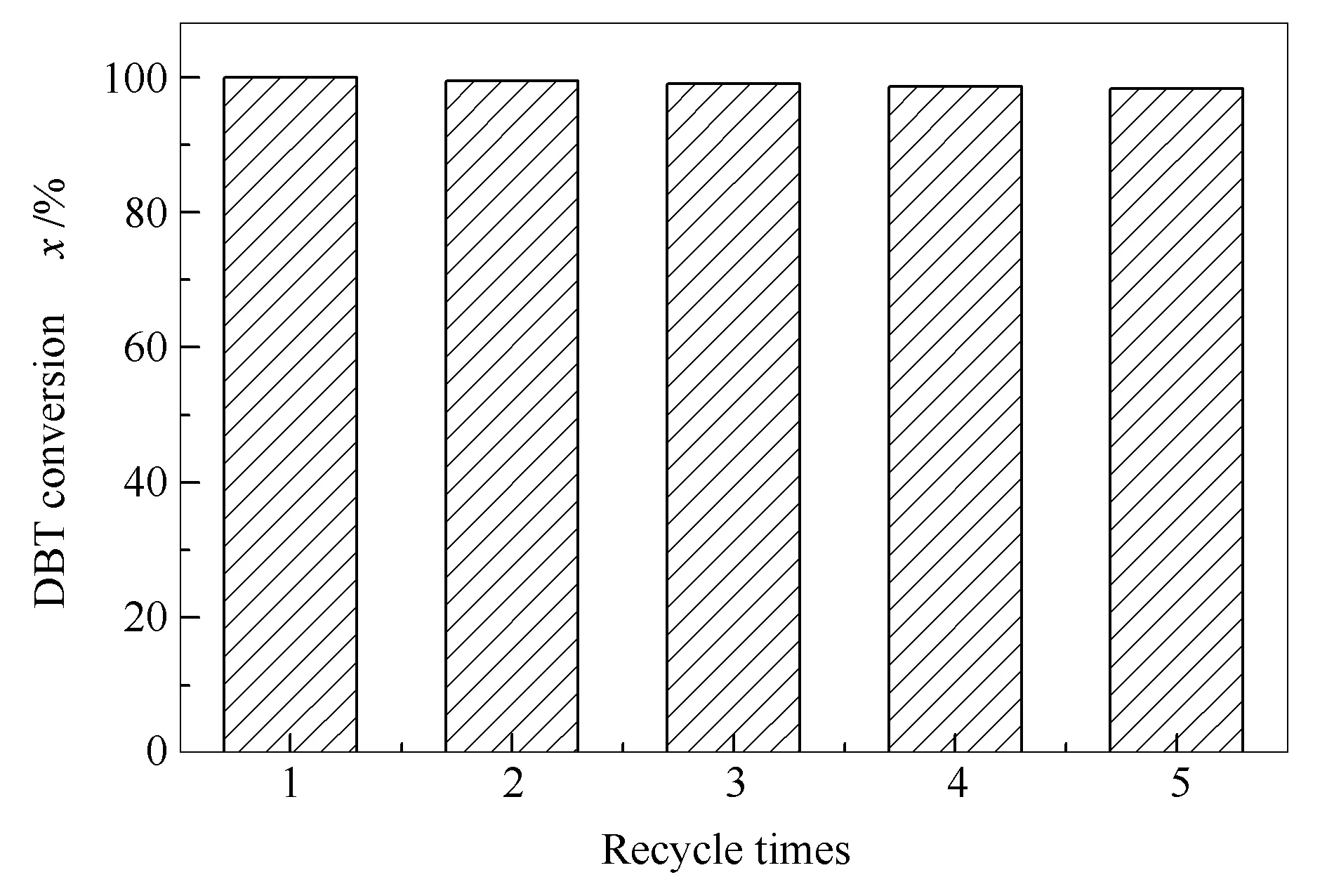
图 9 催化剂重复使用性能的考察
2.3.7 对不同含硫化合物催化氧化脱硫活性的考察
考察了催化剂对苯并噻吩(BT)、二苯并噻吩(DBT)和4,6-二甲基二苯并噻吩(4,6-DMDBT)三种不同含硫化合物的氧化脱硫活性,结果见图10。由图10可知,三种含硫化合物被催化氧化的难易顺序为DBT>BT>4,6-DMDBT。Ostuki等[5]认为含硫化合物中硫原子的电子密度越大,含硫化合物越容易被氧化。BT、DBT和4,6-DMDBT的电子云密度分别为5.739、 5.758和5.760,BT中硫原子的电子密度最小,脱硫转化率最低。4,6-DMDBT的硫原子电子密度虽然比DBT的电子密度大,但其脱硫转化率却较低,这说明在杂多酸季铵盐/H2O2体系中,除了电子因素之外,甲基取代基空间位阻也占据较大作用。
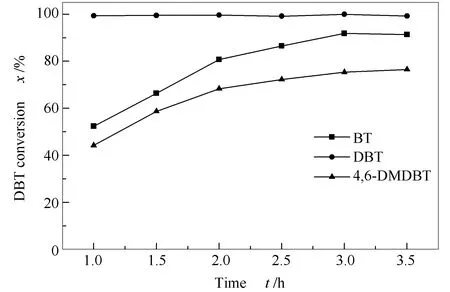
图 10 (CTAB)3H[PW11VO40]催化剂 对不同含硫化合物的氧化脱硫性能Figure 10 Catalytic efficiency in the oxidation desulfurization of BT, DBT and 4,6-DMDBT over (CTAB)3H[PW11VO40] catalyst
3 结 论
采用季铵盐表面活性剂和磷钨钒杂多酸,通过离子交换的方法制备了具有相转移功能的固体催化剂,其中,杂多酸阴离子提供催化活性,季铵盐阳离子提供相转移能力,既提高了催化活性,又便于催化剂的回收再利用。季铵盐的种类和催化剂组成对氧化脱硫活性影响较大,筛选出性能最佳的相转移催化剂为(CTAB)3H[PW11VO40]。红外光谱和XRD表征表明,该催化剂保留了杂多阴离子的结构,且阴离子簇的分布更趋于分散。通过单因素实验法优化了适宜的氧化脱硫反应条件,即n(催化剂)/n(模型柴油)=1∶80,n(H2O2)/n(模型柴油)=8∶1,反应温度50 ℃,反应时间3 h。这种双亲性相转移磷钨钒催化剂,在优化的反应条件下,可以实现100%脱硫,重复使用五次后,催化活性几乎未变。反应后,通过静置破乳,简单过滤即可分离出催化剂和产物。
