乙醇对生物成气过程中煤地质微生物菌群结构及产气途径的影响
2019-11-21杨秀清梁祺韩作颖
杨秀清,梁祺,韩作颖
(1.山西大学 生物技术研究所 化学生物学与分子工程教育部重点实验室,山西 太原 030006;2.煤与煤层气共采国家重点实验室,山西 晋城 048000)
0 引言
煤层气是一种以甲烷为主要成分的自生自储的非常规天然气[1],根据形成机制的不同,分为生物成因气和热成因气两种。随着资源的日益短缺,煤层气越来越受人们的关注,其中生物成因气尤为突出,它的开采和利用使得废弃煤矿等变废为宝,实现了煤层气的再生和资源的再利用。国内外对于微生物增产煤层气的研究涉及范围非常广,如实地勘探、现场注入,实验室条件下模拟产气[2],利用微生物分子生态学的方法进行微生物菌群特征分析等,研究不同成气条件下生物成因气的形成机理及成气过程中的功能微生物菌群。
影响生物成因气生成的因素有很多,主要包括温度、pH、煤的溶解程度和粒径大小、营养物质、盐度、孔隙空间以及沉积速率、沉积环境等。为了增产煤层气,有学者添加一些营养物质、微量元素、维生素等到煤的发酵液中,刺激微生物菌群代谢煤产生甲烷。如Wang 等通过添加酵母膏等营养物质来刺激微生物菌群增加甲烷产量[3], Harris等发现甲酸的添加可以提高甲烷产率[4]。此外,还有研究表明加入新的或外源微生物可增加微生物活性,进而促进煤层气的生成。Jones等用湿地富集的微生物菌群进行模拟产气,发现实验条件下每克煤产生80 μmol甲烷,同时还证实这些微生物菌群有使废弃煤层气井再生产的可能[5]。还有结果表明,改善培养环境可以使微生物更好地和煤进行相互作用。Green等研究了从粉河盆地产出水中富集的产甲烷菌群,发现温度、培养液的pH值以及煤的颗粒度均会对产CH4速率有显著影响[6]。
乙醇作为一种无毒廉价的有机材料而被研究者关注。Bi等通过寻找最佳的培养基配方来增强微生物的活性,他们向培养基中加入一些有机溶剂和表面活性剂SDS,结果表明,添加乙醇、甲醇和异丙醇可以增加煤的生物转化[7]。Zhang 等发现添加乙醇可以提高甲烷含量,且这种作用依赖于乙醇的浓度,当乙醇为100 mmol·L-1时,产甲烷率可以提高24倍,当乙醇为300 mmol·L-1时却没有这种效果[8]。Liu等的实验表明,加入5~10 mg乙醇到10 g煤中,也提高了微生物生产甲烷能力[9]。
本课题组在前期研究中也发现了乙醇可以促进微生物产气,当乙醇含量为1%时,产气量最高,可达44.86 mL·g-1,是不添加乙醇实验组产气量的2倍多。尽管国内外的研究,包括本课题组的研究都证实了添加乙醇可以促进生物产气,但是乙醇为什么会促进煤制甲烷的生成,乙醇的添加与产气微生物之间有什么联系还不得而知。本文在不同的发酵时间取样,通过PCR-DGGE和高通量测序的方法,研究微生物菌群的变化,揭示乙醇在整个发酵过程中导致了哪些微生物的变化及对产气类型的影响,同时也结合实时荧光定量PCR技术来分析微生物菌群数量的变化,以期对发酵过程中微生物菌群结构、数量和产气类型做全方位的分析。
1 材料和方法
1.1 样品采集
煤样取自内蒙古锡林浩特市神华胜利煤田的褐煤。将煤块粉碎过筛后,制成颗粒度为180~250 μm(60~80目)的煤粉,80℃真空干燥24 h,放入干燥器中备用。实验所用接种物为山西寺河矿区121煤层气井中的产出水样富集驯化后的培养液。
1.2 主要试剂和仪器
DNA胶回收试剂盒、Proteinase K,生工生物工程股份有限公司;十二烷基硫酸钠(SDS),北京索莱宝科技有限公司;EasyTaq ploymerase,北京全式金生物技术有限公司; 10 000×4s Red plus、过硫酸铵(APS)、四甲基乙二胺(TEMED)、丙烯酰胺、尿素、甲酰胺、双丙烯酰胺,生工生物工程股份有限公司;机械振荡器Mini-Bead Beater,BioSpec Products,USA;PCR仪,美国伯乐公司PTC-200型;酶标仪,美国伯腾仪器有限公司;YQX-Π 型厌氧手套箱,上海跃进医疗器械厂;电泳仪,北京六一仪器厂;美国伯乐DCodeTM通用突变检测系统,美国BIO-RAD 公司;Gel Doc 2000 UV 凝胶成像系统,美国BIO-RAD 公司。
1.3 实验用培养基
基础培养基配方(g·L-1):酵母抽提物2.0 g,K2HPO42.9 g,KH2PO41.5 g,NH4Cl 1.8 g,MgCl20.4 g,半胱氨酸 3 g,刃天青0.002 g。
微量元素溶液配方为(g·L-1):氮三乙酸 1.5 g,CaCl20.1 g,MgSO4·7H2O 3.0 g,H3BO30.05 g,FeSO40.1 g,NaCl 1.0 g,COCl20.1 g,MnSO40.5 g,ZnSO40.1g,NaMO40.05 g,A1K(SO4)20.01 g,NiCl20.1g,CuSO40.01 g。
1.4 厌氧发酵实验
以500 mL的厌氧瓶作为反应容器,加入250 mL培养基,50 mL菌液,及20 g煤粉,将实验分为3组,每组设置3个重复。第1组添加1.5 mL乙醇(0.5%,V/V),第2组添加3.0 mL乙醇(1%,V/V),对照组不加乙醇,其余条件都相同。以上操作全部在厌氧操作箱中进行。
1.5 基因组DNA的提取
在第28、35、42、49、60、86和92 d(依次命名为28、35、42、49、60、86、92)分别进行取样。乙醇含量0%、0.5%、1%,分别命名为A,B和C三组。将采集的各样品于12 000 r/min条件下离心1 min,收集菌体供提取煤地质微生物基因组。提基因组的方法参照杨秀清等[10]。收集的菌体经TPM、STE洗涤后,用Mini-Bead Beater (MBB)玻璃珠进行细胞破碎(2 500 r/min,30 s),2~3。合并上清液于新的EP管中,加入SDS、蛋白酶K对菌体进行裂解(55℃,2~3 h)。再用有机溶剂酚-氯仿,异丙醇进行抽提,最后用75%的乙醇进行洗涤,加ddH2O溶解。用0.7%琼脂糖凝胶电泳检测所提基因组的大小。用酶标仪检测所提基因组的浓度与纯度。
1.6 16SrRNA基因片段的扩增
提取的基因组DNA作为模板,以细菌V3区(338F/518R)和古菌V3区(0357F/Arch519R)的通用引物进行目的片段扩增。PCR扩增体系:10×Easy Taq buffer 5 μL,dNTPs(2.5 mmol/L)4 μL,10 μmol/L的上下游引物各2 μL,Easy Taq ploymerase 1 μL,DNA模板2 μL,加蒸馏水至50 μL。PCR反应程序:95℃ 5 min;95℃ 30 s,55℃ 30 s,72℃ 45 s,30 个循环;72℃ 10 min。细菌及古菌PCR扩增体系及反应程序相同。PCR产物在1.5% 的琼脂糖凝胶中检测目标片段长度,以验证PCR的正确性,随后用胶回收试剂盒对目的产物进行纯化,供高通量测序使用。DGGE上样所需PCR产物以富含GC含量的引物(细菌338F-GC/518R,古菌0357F-GC/Arch519R)进行PCR。
1.7 DGGE
PCR-DGGE分析使用美国BIO-RAD 公司的D-code 系统。将细菌PCR 产物上样到质量分数10%聚丙烯酰胺凝胶上,古菌PCR 产物上样到8%聚丙烯酰胺凝胶上,60℃ 85 V下电泳12 h,变性梯度范围为40%~60%。电泳完毕后,将胶放入3×的4s Red plus 染液中染色20 min 左右,用凝胶成像仪记录图像,并用QuantityOne 软件进行分析。
1.8 实时荧光定量PCR
PCR反应体系为20 μL,上下游引物各0.8 μL,SYBR Green PCR Master Mix 10 μL,ddH2O 7.3 μL,Roxdye 0.1 μL,基因组DNA模板1 μL。细菌所用引物为(338F/518R),古菌所用引物为(344F/Arch519R)。
PCR反应程序:95℃ 5 min;95℃ 10 s,55℃ 30 s,30 个循环。每个循环结束后,收集荧光信号。本实验采用Bio-Rad CFX Manager进行实验结果的记录与分析,每个样品重复3次,最后结果取平均值。
统计学方法:选用SPSS 22.0软件进行统计学分析,两组间的比较用独立样本t检验,α=0.05为检验标准。
1.9 高通量测序数据处理
测序完成后,使用 FLASH v1.2.7软件,对每个样品的 reads 进行拼接;然后使用 Trimmomatic v0.33软件,对拼接得到的 Raw Tags进行过滤,得到高质量的 Tags 数据;最后使用 UCHIME v4.2软件,鉴定并去除嵌合体序列,得到最终有效优质序列。用QIIME(version 1.8.0)软件中的 UCLUST对优质序列在97%的相似度水平下进行聚类、获得OTU,并进行物种注释及丰度分析。基于OTU数量的结果,评估样品生物α多样性(包括 Chao1 值、ACE 值、Shannon 以及Simpson指数)和β多样性。
2 结果与分析
2.1 PCR-DGGE分析
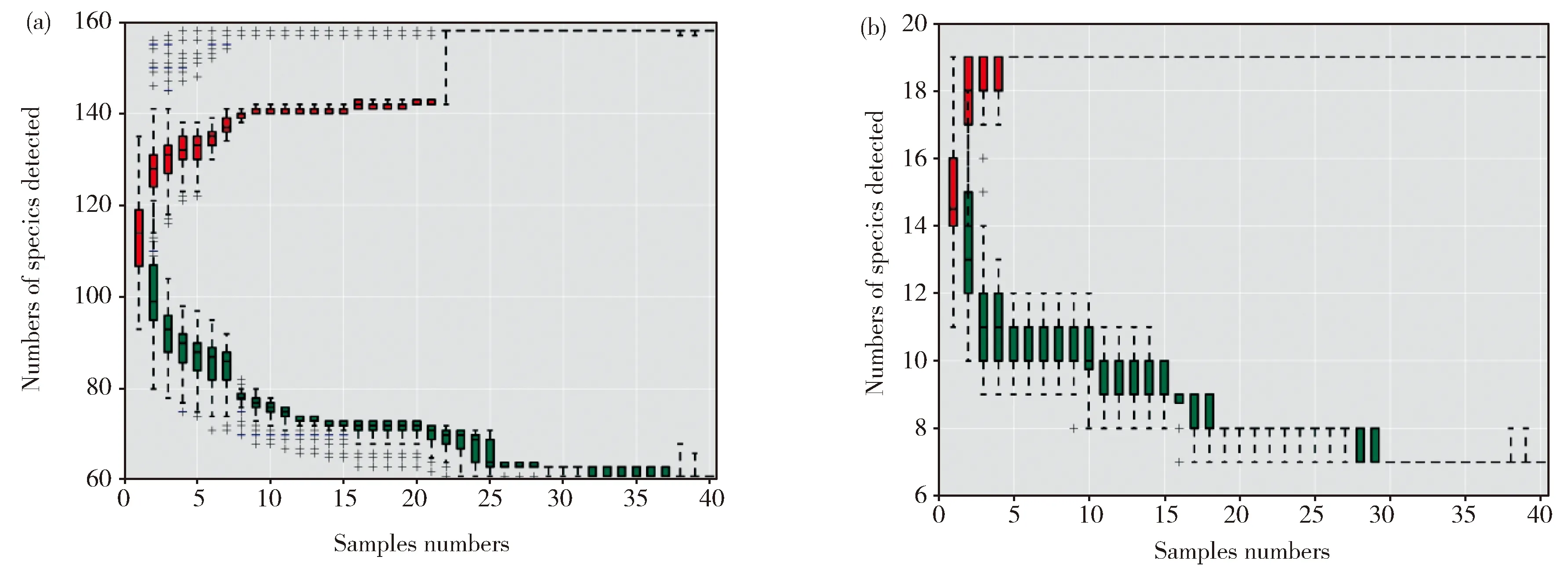
细菌的16S rRNA V3区PCR-DGGE图谱见图1,古菌的16S rRNA V3区PCR-DGGE图谱见图2,所用Marker参照杨秀清等制备[10-11]。样品经过变性梯度凝胶电泳后,分离出数量不等的条带,细菌图谱中各个样品中有3~15条条带,古菌图谱中各个样品有2~4条条带,可见各个样品中细菌物种较为丰富,古菌物种则较少。各个样品有共同的条带,但是条带的亮度不同,表明微生物菌群的丰度不同。样品中大多数条带都与细菌Marker一致,这些条带所对应的菌种在整个发酵过程中起主导作用。与物种Marker对比可知,细菌图谱中所对应的条带分别属于变形杆菌属(Proteus)、脱硫弧菌属(Desulfovibrio)、屠场杆状菌属(Macellibacteroides)、柠檬酸杆菌属(Citrobacter)、梭菌属(Clostridium)、Parabacteroides,古菌图谱中的条带分别属于甲烷杆菌属(Methanobacterium)、甲烷八叠球菌属(Methanosarcina)。用Quantity one软件对DGGE图谱进行优化处理,即基本的背景排除功能,然后经过泳道(Lane)识别、条带(Band)识别和配对,对图谱中的条带进行定量分析。细菌及古菌的菌群相对含量柱状图见图3和图4。根据图3可以看出不同样品中细菌的种类较多,各菌群的相对丰度变化较小,不会由于乙醇含量的增加而发生明显的改变。而图4显示仅有两种甲烷古菌物种,即Methanosarcina和Methanobacterium。它们随着样品的变化,呈现一个较大的波动。随着样品从A组到B、C组,Methanobacterium出现一个先减少后增多的现象,而Methanosarcina则是先增多后减少。在A49中Methanosarcina所占比例为60%,而Methanobacterium仅占40%。在B42中,Methanobacterium占优势(72%),而Methanosarcina则占28%,在C42中,出现同样的现象。可见,乙醇的添加对产甲烷古菌有明显的影响。
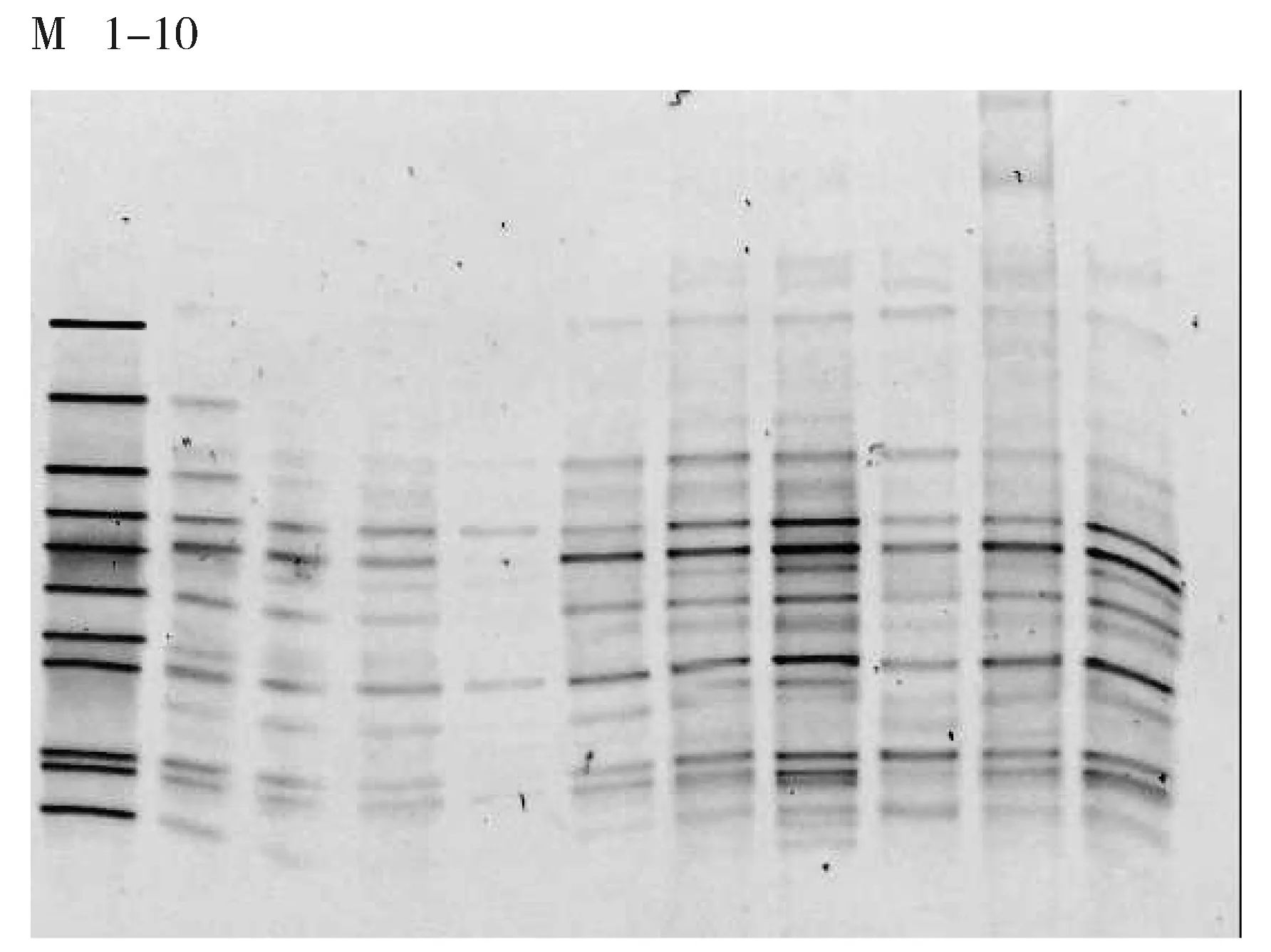
M:Marker.It represented Clostridium, Enterococcus, Parabacteroides, Macellibacteroides, Proteus, Desulfovibrio,Raultella Citrobacter, Cloacibacillus, Stenotrophomonas, Acetoanaerobium, respectively from top to bottom;1-10 represented samples for A28, A35, A42, A49, B28, B35, B42, C28, C35 and C42 respectivelyFig.1 PCR-DGGE fingerprints of 16S rRNA V3 region of bacteria in some samplesM:Marker 从上到下依次为Clostridium、Enterococcus、Parabacteroides、Macellibacteroides、Proteus、Desulfovibrio、Raultella Citrobacter、Cloacibacillus、Stenotrophomonas、Acetoanaerobium;1-10依次为样品 A28、A35、A42、A49、B28、B35、B42、C28、C35、C42图1 部分样品细菌的16S rRNA V3区PCR-DGGE指纹图谱
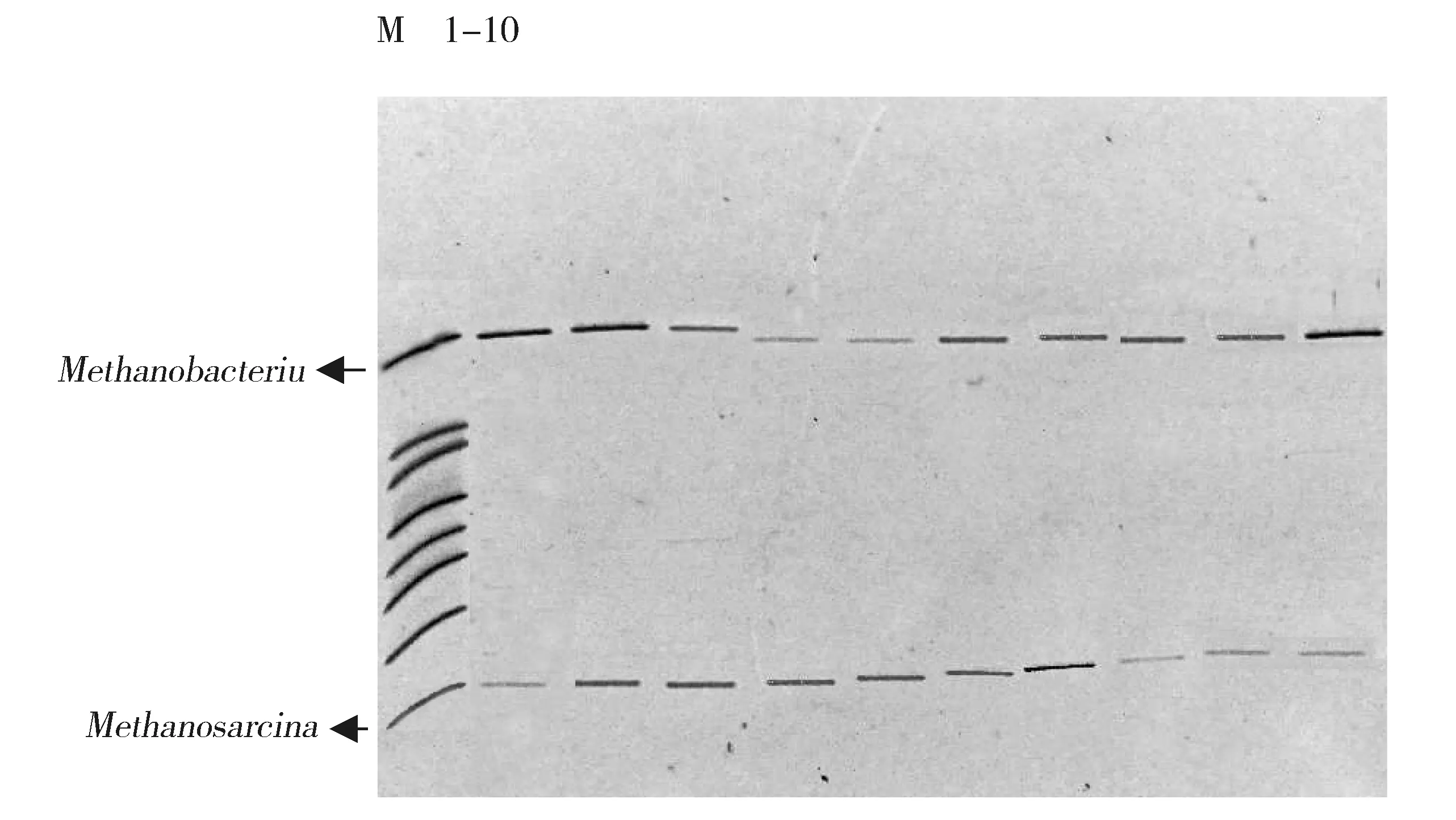
M: Marker 1-10 represented samples for A28, A35, A42, A49, B28, B35, B42, C28, C35 and C42 respectivelyFig.2 PCR-DGGE fingerprints of 16S rRNA V3 region of archaea in some samplesM:Marker 1-10依次为样品A28、A35、A42、A49、B28、B35、B42、C28、C35、C42图2 部分样品古菌的16S rRNA V3区PCR-DGGE指纹图谱
2.2 实时荧光定量PCR
各样品中细菌及古菌的数量见表2。从2表可以看出,发酵天数为28 d和35 d时,A、B、C各组不论是细菌还是古菌的菌群数量之间不存在明显差异(P>0.05),说明在短期发酵时间内,乙醇的作用发挥的不明显。当发酵天数为42 d、49 d和60 d时,B、C组与A组菌群数量之间存在明显差异(P<0.05),说明随着发酵时间的延长,乙醇开始发挥作用,菌群数量开始明显增加。总的来讲,随着发酵时间的延长,菌群数量不断增加,乙醇的添加导致了微生物菌群数量的明显增加,但是它作用的发挥还受发酵时间的影响。细菌的菌群数量要多于古菌的菌群数量,由于产甲烷古菌只能利用像H2/CO2、甲酸、乙酸等简单的物质来产甲烷,所以推测乙醇是被细菌微生物直接利用,导致细菌菌群较为活跃,细菌菌群数量多且增长快。
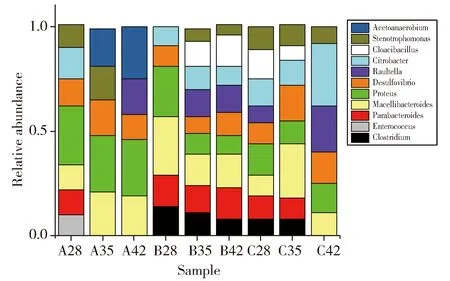
Fig.3 Relative abundance histogram of bacterial flora in DGGE spectrum图3 DGGE图谱细菌菌群相对丰度柱状图

Fig.4 Relative abundance histogram of archaea flora in DGGE spectrum图4 DGGE图谱古菌菌群相对丰度柱状图
2.3 高通量测序分析
高通量测序结果显示,细菌40个样品测序共获得2 522 076条优质序列,古菌40个样品测序共获得2 311 412条优质序列。将序列之间相似性高于97%定义为一个OTU,每个OTU对应于一种代表序列。细菌菌群OTU统计为283,古菌菌群OTU统计为22。
图5a和图5b为细菌和古菌属水平物种累积曲线图,反映了样本数量与注释到的物种数量间的关系。单个红色箱形反映了抽取的样本的总种数;总的红色箱形组成了累积曲线,反映了持续抽样下新物种出现的速率。单个绿色箱形反映了抽取的样本中的种数;总的绿色箱形组成共有量曲线,反映了持续抽样下样本共有种出现的速率。在一定范围内,随着样本量的加大,若曲线急剧上升,则代表群落中有出现大量新种,说明样本量不足。若曲线趋于平缓,则表示抽样充分。从图5a和图5b可以看出,不管是总种数还是共有种数,随着样品数的增加,都呈现平缓的趋势。可见我们样本量充分,可以进行数据分析。
2.4 序列号
研究得到的高通量测序结果提交至NCBI SRA (Sequencet Read Archive)数据库,收录号为SRS3948544。
2.5 产甲烷混合菌群群落结构及动态变化
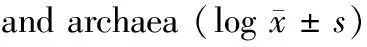

样本名称细菌数量/mL古菌数量/mL A 283.02±0.782.22±0.39 B 284.71±0.583.78±0.26 C 283.50±0.353.55±0.42 P值0.210.32 A 353.66±0.402.55±0.31 B 353.06±0.643.83±0.25 C 353.72±0.223.71±0.32 P值0.100.22 A 423.85±0.57a3.66±0.39a B 425.67±0.45b3.81±0.56b C 424.78±0.27b3.71±0.45b P值0.030.03 A 493.28±0.65a3.73±0.12a B 494.63±0.36b3.94±0.47b C 494.79±0.32b4.79±0.50b P值0.030.02 A 603.63±0.60a3.77±0.48a B 605.41±0.55b4.88±0.36b C 605.87±0.39b5.08±0.21b P值0.010.01 注:同列上角标相同字母表示差异不显著(P>0.05),不含相同字母表示差异显著(P<0.05)。
The red box represented the total number of species containing in the sampled samples,and the green box represented the number of species in common in the sampled samplesFig.5 (a) Species accumulation curve of bacteria genus;(b) Species accumulation curve of archaea genus红色箱形代表了抽取的样本中所含有的总物种数,绿色箱形代表了抽取的样本中所共有的物种数图5 (a)细菌属水平物种累积曲线图;(b)古菌属水平物种累积曲线图
为了便于数据分析,随机挑选了一些样品的测序结果进行展示。如图6a所示,在产气发酵过程中,微生物细菌群落在门的水平上主要由厚壁菌门 (Firmicutes)、变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)组成。如图6b,从细菌属水平上群落丰度分析,主要的菌属有变形杆菌属、脱硫弧菌属、屠场杆状菌属、Paraclostridium、柠檬酸杆菌属,还有少量的肠球菌属(Enterococcus)以及Tyzzerella。整体来看,随着发酵时间的延长,A、B、C三个实验组的物种趋势的变化是相似的。对于古菌菌群,主要是广古菌门(Euryarchaeota);从属的水平分析(图7b),主要由Candidatus-Methanoplasma、甲烷杆菌属、甲烷八叠球菌属组成,还有少量的产甲烷袋菌属(Methanofollis)。值得注意的是,在古菌门和属的物种分布图中出现了细菌的身影,这是由于细菌的16S序列与古菌的16S序列有一定的相似性,所以在进行高通量测序时,虽然使用古菌特异性引物扩增的16s序列,但是还是不可避免的非特异性扩增出细菌的16s序列[12]。在Yang等人的研究中也出现类似的情况[13]
在样品A28中,Candidatus-Methanoplasma占绝对优势(达81.75%)。随着发酵时间的延长,Candidatus-Methanoplasma越来越少,在发酵60 d后,Candidatus-Methanoplasma减少为13.66%(A60)。A、B、C三组样品中,随着发酵时间的延长,Candidatus-Methanoplasma都有所减少。
在A组样品中,A28中Methanosarcina含量为4.33%,A49中Methanosarcina含量为7.16%,A60中Methanosarcina含量为46.55%,随着发酵时间的延长,Methanosarcina逐渐变为优势菌属。而在B、C组样品中,Methanobacterium逐渐成为优势菌属,B92中Methanobacterium含量为46.80%,C92中Methanobacterium含量为64.92%。表明,乙醇的添加对细菌微生物菌群结构影响不大,但明显改变了产甲烷古菌的微生物菌群结构,与PCR-DGGE结果一致。
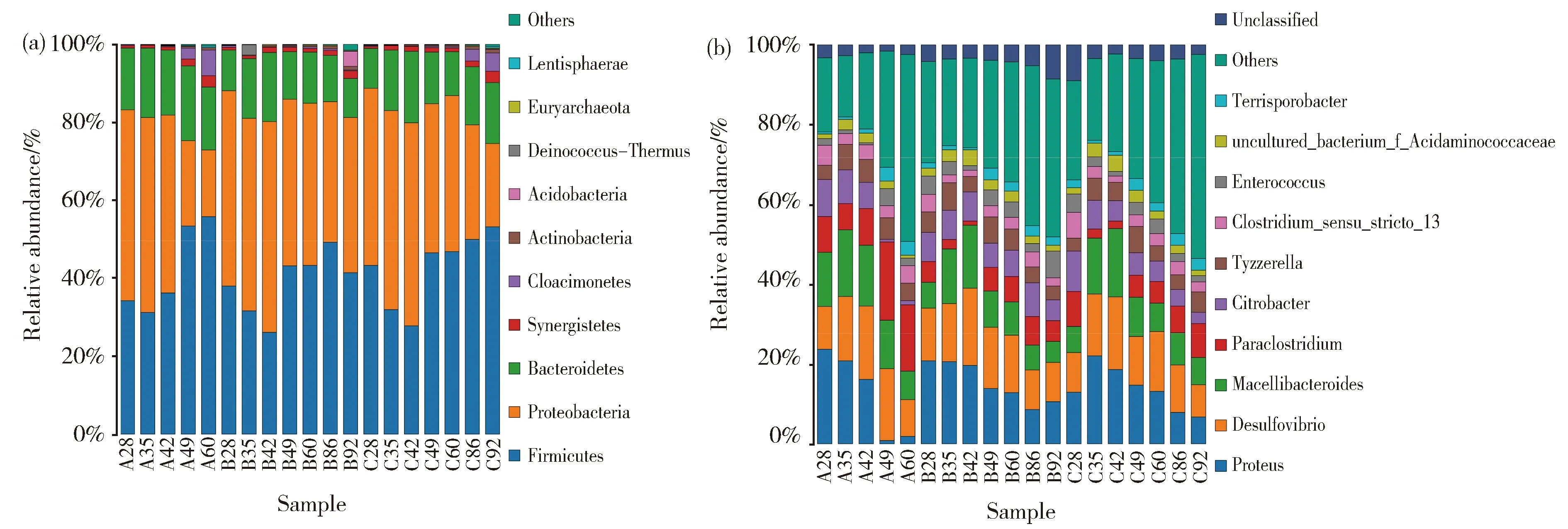
Fig.6 Bar chart of the relative abundance of the top 10 bacteria of each group in (a) phylum and (b) genus level图6 细菌门水平(a)和属水平(b)的物种分布图
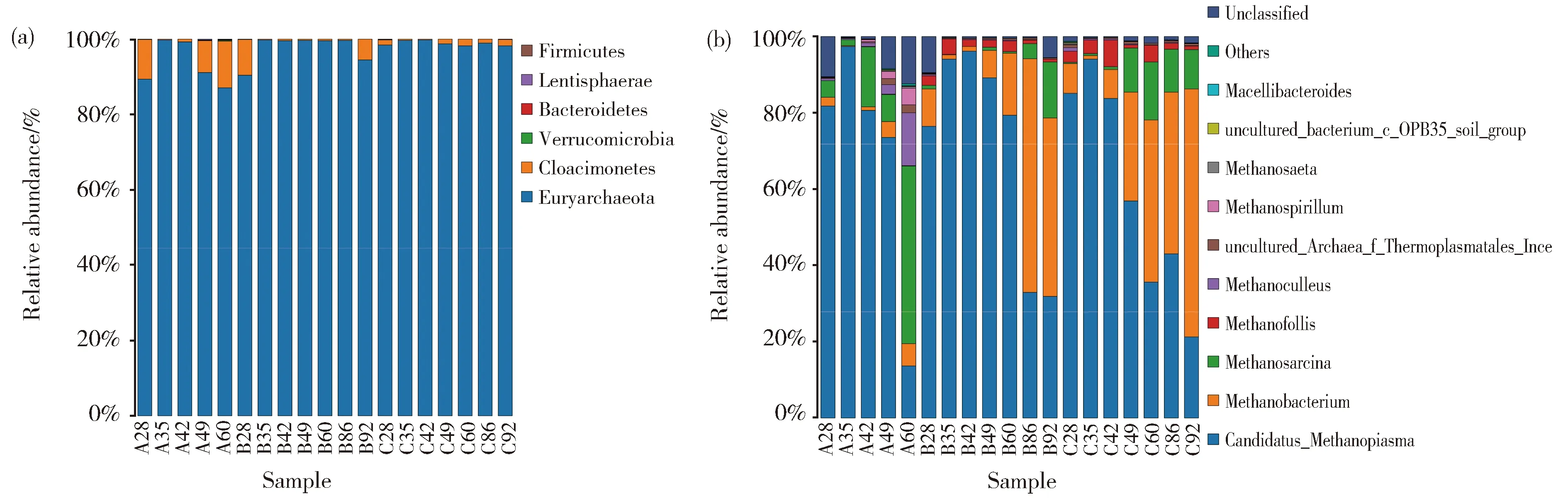
Fig.7 Bar chart of the relative abundance of the top 10 archaea of each group in (a) phylum and (b) genus level图7 古菌在门水平(a)和属水平(b)的物种分布图
2.6 产气途径分析
根据产甲烷菌的产甲烷机制不同,将生物成因气的产气途径分为三大类:(1) 氢营养型,将CO2还原转化为甲烷;(2) 乙酸营养型,转化乙酸产生甲烷;(3) 甲基营养型,转化甲基类化合物如CH3OH、C13H13N、C2H6S等生成甲烷。在煤层气田中,氢营养型产气途径最为常见,乙酸营养型次之,甲基营养型最少。本研究中PCR-DGGE与高通量测序结果显示,在发酵初期,Candidatus-Methanoplasma占优势,该菌属于混合营养型产气[14]。随着发酵时间的延长,对照组中Methanosarcina增多,甲烷产量增加;添加乙醇的实验组则是Methanobacterium逐渐增多。Methanosarcina可利用广泛的底物产甲烷,但在煤层气的研究中以降解乙酸为主[15],所以对照组的产气类型为乙酸营养型。而Methanobacterium则是典型的氢营养型产甲烷菌属[16],故添加乙醇的实验组产气类型为氢营养型。可见乙醇的加入导致了本实验中甲烷产气类型的而改变,由混合型和乙酸营养型的产气类型改变为氢营养型的产气类型。
3 讨论和结论
微生物增产煤层气被认为是具有应用前景的一种煤层气再生方式,可以有效缓解能源短缺。国内外的研究者通过从微生物刺激、微生物强化、增加煤溶解度和生物利用率等方面来增产煤层气,特别是在添加营养物质刺激微生物复苏和生长,已成为研究的热点[17]。煤地质微生物处在一种较为恶劣和缺乏营养的环境下,处于不活泼的状态,需要某些环境的刺激,才能更好地进行代谢活动。乙醇作为一种绿色、环保、廉价的有机物,也被用于微生物增产煤层气的研究。通过添加乙醇来刺激微生物活性,发现添加乙醇可以增加甲烷的产量[7-9],证实了利用乙醇可以增产煤层气,因其无毒在现场实验中便于操作,不需要繁杂的防护措施;还因其廉价,可以在实地进行大规模的实验研究,其增产煤层气产生的经济效益远大于乙醇的投入成本。所以利用乙醇增产煤层气具有一定的可行性和经济效益。但是在产气过程中,微生物菌群如何变化,与乙醇的添加有何关联还有待进一步研究。
本文通过PCR-DGGE、实时荧光定量PCR、高通量测序相结合的方法,研究了在乙醇存在下,生物成气过程中微生物菌群结构和数量的变化。结果显示,随着发酵时间的延长,样品中细菌及古菌菌群数量不断增加。细菌菌群结构随着乙醇的添加变化不明显,而古菌菌群结构则受乙醇的强烈影响。高通量测序结果显示,细菌物种组成在门水平上主要有三大类,分别是厚壁菌门、变形菌门、拟杆菌门,与PCR-DGGE指纹图谱显示的结果相同。从细菌属的分类学水平来看,主要参与的菌属有变形杆菌属、脱硫弧菌属、屠场杆状菌属、梭状芽胞杆菌属、柠檬酸杆菌属(Citrobacter),还有少量的肠球菌属以及Tyzzerella。PCR-DGGE中显示条带所代表的物种主要有Proteus、Desulfovibrio、Macellibacteroides、Citrobacter、Clostridium、Parabacteroides。二者显示结果几乎一致。比较这二者的物种相对丰度柱状图可知,物种组成及变化趋势是相同的,即乙醇的添加对细菌菌群结构变化影响不大。实时荧光定量PCR结果显示,细菌微生物菌群的数量随着发酵时间的延长而不断增加。这些细菌微生物菌群属于发酵细菌和产氢产乙酸菌,它们共同作用参与大分子煤的降解过程。厚壁菌门中的微生物主要参与一些混合酸、醇和中性物质的生成,其中的梭菌科在一些淀粉、纤维质、几丁质等的解聚中扮演重要角色[16],样品混合菌群中属于该门的菌属有Paraclostridium、Tyzzerella、Enterococcus。变形菌门是种类较为丰富的一类细菌。一般生物成因煤层气中的变形菌主要以互养型的β-,γ-和δ-变形菌为主[16],Desulfovibrio属于变形菌门的δ-变形菌,是一种硫酸盐还原菌,仅能利用硫酸盐进行呼吸作用,其次,Deslfovibrio也可以和产甲烷古菌一样能够利用乙酸和H2,从而进而促进煤大分子的降解[18]。Proteus是一种兼性厌氧的革兰氏阴性细菌,以氧化和发酵形式分解糖类,产酸产气。拟杆菌门在沉积物中较常见,是一大类化能自养型微生物,样品混合菌群中属于该门的菌属有Macellibacteroides、Citrobacter,它们主要参与大分子如蛋白质、糖、纤维素等的降解,发酵成为甲酸、氢气和二氧化碳[19-20]。
高通量测序得出,古菌物种组成在门的水平上主要是广古菌门,与PCR-DGGE结果完全一致。从属的水平看,主要由Candidatus-Methanoplasma、甲烷杆菌属、甲烷八叠球菌属组成。PCR-DGGE指纹图谱所示条带有两条,分别为Methanobacterium、Methanosarcina,没有出现Candidatus-Methanoplasma,可能是由于Candidatus-Methanoplasma属下有很多的种,高丰度的属分散在不同的种里,丰度不会那么集中,而PCR-DGGE指纹图谱只能显示属水平上的丰度,所以受到干扰显示不出来,具体原因还需要进一步研究。Candidatus-Methanoplasma属于Methanomassiliicoccales,这是迄今为止发现的第七类产甲烷菌目[21],是由专性的氢依赖性甲基营养菌组成,属于混合营养型[14]。本研究使用的接种物与杨秀清等[22]研究中使用的接种物相同,然而其研究中并未发现Candidatus-Methanoplasma。在煤层气微生物菌群的研究也几乎没有发现过这种细菌,而是在反刍动物和人类粪便的胃中发现有它的身影[21,23],这种古菌并不利于甲烷的生成。本研究中使用的接种来源是富集和驯化后的培养基,推测这种古菌是在富集和驯化过程中产生的。比较PCR-DGGE和高通量测序二者的物种柱状图,物种组成及变化趋势是相似的,添加乙醇的实验组与对照组之间存在明显的差别。对照组中,随着发酵时间的延长,甲烷八叠球菌属逐渐增多,添加0.5%和1%乙醇的实验组中则是甲烷杆菌属明显增加。Methanosarcina具有广泛的可利用底物,能够利用乙酸、CO2、甲醇、甲胺、甲基硫化物等多种有机或无机化合物为底物产生甲烷[24]。但在有关煤开采研究中,降解乙酸是主要途径,因为乙酸盐比氢气更易获得[15],因此Methanosarcina产甲烷途径应该是乙酸型,而Methanobacterium则是典型的氢营养型产甲烷菌属[15]。所以推测随着发酵时间的延长,对照组以混合营养型和乙酸型的产甲烷途径为主,添加乙醇的实验组以氢营养型产甲烷途径为主。
