多位点序列分析方法在克罗诺杆菌属菌株溯源分析上的应用
2019-11-07闫瑞杨捷琳陈翠玲钮冰徐之雯蒋原
闫瑞,杨捷琳,陈翠玲,钮冰,徐之雯,蒋原
(1.上海大学生命科学学院,上海 200444;2.上海出入境检验检疫局,上海 200135)
0 引言
克诺罗杆菌原为阪崎肠杆菌(Enterobacter sakazakii),隶属于肠杆菌科[1-2]。克罗诺杆菌属(Cronobacter spp.)现包括7个种和3个亚种[1-3],属内菌株不同分型种类与临床相关性存在较大差异,其中多数临床分离株为阪崎克罗诺杆菌(C.sakazakii)和丙二酸盐阳性克罗诺杆菌(C.malonaticus)。据报道,多数菌株易感染婴幼儿和老年人等免疫力低下人群,可导致严重疾病(坏死性小肠结肠炎、败血症和脑膜炎),且致死率非常高(40%~80%)[4-6]。随着克罗诺杆菌在不同食品尤其是婴幼儿配方粉中被检出而受到食品和卫生监管部门的高度重视[7,8]。
尽管人们已从食品、环境和临床上分离到大量菌株,也对其分类有了较为清晰的认识。但是随着越来越多克罗诺杆菌全基因组测序的完成,研究发现过去基于单个或多个基因的分型方法由于纳入分析的信息有限,误将许多无关菌株划分到了一起[9,10]。因此利用全基因组测序(W hole genome sequencing,WGS)获得的基因组数据,采用合适的分型方法来准确鉴定和快速识别不同来源的克罗诺杆菌能够更为精确的将菌株进行分类,全基因组所提供的信息量在分类学中就显得尤为重要。目前,已有许多方法可以分析全基因组测序数据,常用的3种方法[11]包括:基于k-m er统计的分析方法,单核苷酸多态性分析(Single nucleotide po ly-m orphism s,SNP)以及基于W GS的扩展的多位点序列分型方法即全基因组MLST。其中全基因组MLST具有较高的流行病学一致性、稳定的菌株命名法和完善的数据库,能够实现国际标准化和可扩展性,分析速度快、辨识度高,计算要求复杂度低且相较于另外两种分析方法无需生物信息专业知识。正是由于全基因组MLST具备上述诸多优势,使其成为国际食源性疾病监测分子分型网络(PulseN et International,http://www.pu lsenetinternational.org/)标准化监测的技术基础[11-12]。
本研究中所采用的扩展的多位点序列分型方法是由Forsythe等[9]人建立的cogMLST分析方案,该方案主要是以C.sakazakii ES15菌株的直系同源基因簇作为参考基因组所定义的1865个基因座为基础进行基因组序列比对分析,可以跨属或选择特定的物种或菌株进行比较。2014年Forsythe等[9]通过采用更具辨别力的编码核糖体蛋白基因的多位点序列分析(ribosom al-MLST,rM LST)和基于直系同源基因簇核心基因的多位点序列(C lusters o f O rtho logous Genes-core geneM LST,COG-cgM LST)分析方法对107个克罗诺杆菌全基因组数据分析进一步证明C.sakazakii CC 4克隆谱系非常稳健。现在cogMLST可通过克罗诺杆菌PubM LST数据库在线进行全基因组测序数据分析。
1 材料与方法
1.1 数据来源
克罗诺杆菌属菌株信息收集自http://pubm lst.org/cronobacter/,通 过 Cronobacter Pub MLST门户网站https://pubm lst.org/bigsdb?db=pubm lst_cronobacter_isolates,点击查询/浏览数据库(Search or brow se database)选项下的基于7个位点的MLST方案获取克罗诺杆菌的序列类型并将所有菌株来源信息以Excel文件格式导出,后通过Excel工作表中的数据筛选功能对菌株进行统计分析。
本研究中使用的菌株信息均来自C ronobacter Pub MLST数据库中的2 438株克罗诺杆菌属菌株,包括阪崎克罗诺杆菌ST 1(67株),ST 4(95株),ST 13(43株)和丙二酸盐阳性克罗诺杆菌ST 7(35株),具体信息见表1。表2列出了阪崎克罗诺杆菌和丙二酸盐阳性克罗诺杆菌中主要序列类型的国家来源概况。
1.2 建立本地数据库
全基因组序列分析所需的240株菌株DNA序列收集自http://pubm lst.org/cronobacter/,通过C ronobacter PubM LST门户网站上的选项进行计算机分析,可访问:https://pubm lst.org/bigsdb?db=pubm lst_cronobacter_isolates。浏览并选取已完成全基因组测序的菌株以FASTA文件格式导出菌株的DNA序列信息。
1.3 7-loci MLST
菌株(2 400多株)的序列类型和克隆谱系通过Cronobacter Pub-MLST(https://pubmlst.org/cronobacter/)的在线工具进行确定,访问该网站并选择序列比对(Sequence query)选项下的MLST方案即获得相应的等位基因编号,ST和CC等比对信息。
1.4 cog MLST分析
首先通过Cronobacter Pub MLST(https://pubmlst.org/bigsdb db=pubmlst_cronobacter_isolates)当中的“分析”(Analysis)选项下的基因组比较(Genome Comparator)选项进行提交所需分析菌株的完整DNA序列,后选用cog MLST方案依次重复获取阪崎克罗诺杆菌ST1(67株),ST4(95株),ST 13(43株)和丙二酸盐阳性克罗诺杆菌ST7(35株)菌株基因组的距离矩阵。矩阵以nexus文件格式导出并使用SplitsTree 4中的默认设置进行分析(UncorrectedP用于计算距离,并通过Neighbor Net构建网络图谱)。
2 结果与分析
2.1 克罗诺杆菌属菌株统计结果
目前Cronobacter Pub MLST基因组和序列定义数据 库(http://pubmlst.org/cronobacter/)已 包 含 超 过2400多株菌株分离信息,其中C.sakazakii(1 774株)和C.malonaticus(295株)为该属的优势菌种(表1)。通过将7-loci MLST应用于2 438株菌进行分析,共揭示了591种可定义的序列型(sequence type,ST),其中克诺罗杆菌C.sakazakii ST1(280株)、ST 4(334株)、ST 13(67株)和C.malonaticus ST7(78株)为主要序列型(表2)。截止2019年3月20日,数据库中已有623株菌完成全基因组测序,包括C.sakazakii(438株)、C.malonaticus(82株)、C.dublinensis(56株)、C.turicensis(23株)、C.muytjensii(13株)C.universalis(9株)和C.condimenti(2株)。数据库中最早的分离株为1950年分离自英国奶粉中的C.sakazakii,所有菌株共分离自全球40多个国家和地区。多数菌株分离自食品及配料当中,C.sakazakii和C.malonaticus与临床及婴幼儿配方奶粉密切相关。C.sakazaki i和C.malonaticus中主要序列类型的国家来源统计结果显示,多数菌株分离自中国和美国(表1、2)。上述结果表明,由于MLST所针对的七个基因位点在基因组总量中占比较小,导致同一ST型中包含地理来源,分离时间,宿主等不相关的菌株,而对菌株溯源结果适得其反。

表1 Cronobacter Pub MLST数据库中的克罗诺杆菌菌株统计
2.2 C.sakazakii ST 1菌株的cog MLST分析
如图1所示,67株阪崎克罗诺杆菌ST 1菌株的cog MLST聚类分析结果显示,多数ST 1菌株基因组存在较大差异,22株于2014年分离自美国威斯康星州环境中的菌株聚集到了一起。值得注意的是,同样分离自于2014年分离自美国俄勒冈州环境中的两株菌(编号1344和1361)形成了另一分枝。67株ST 1菌株中最早的分离株(编号2522)在1987年分离自丹麦临床当中与两株分别源自爱尔兰和加拿大临床的菌株(编号1912和2536)聚集到了一起。两株最新于2017年分离自丹麦临床的菌株(编号2521和2573)却与最早分离株(编号2522)存在较大差异。这些结果表明,阪崎克罗诺杆菌ST 1菌株的cogMLST聚类与菌株来源之间存在密切相关性。
2.3 C.sakazakii ST 4菌株的cog MLST分析
通过将cog MLST方案应用于95株阪崎克罗诺杆菌ST4菌株进行全基因组分析,结果如图2所示,2014年分离自法国临床及婴幼儿配方奶粉的15株菌都聚集到了一起,3株源自2009年美国临床的菌株(编号121、1163和1315)形成了一个单独分枝。此外,一株于1997分离自年荷兰临床的菌株(编号1104)与另一株于2009年分离自瑞士临床的菌株(编号2539)聚集到了一起。另外,最早于1950年分离自英国奶粉 的两株菌则未与其它分离菌株形成明显聚类。95株阪崎克罗诺杆菌ST 4菌株的cogMLST聚类分析结果表明,相同序列型菌株包含了大量非相关菌株,聚类与菌株来源之间存在一定关联。

表2 C.sakazakii和C.malonaticus中主要序列类型的国家来源概况

图1 C.sakazakii ST1菌株的cog MLST分析
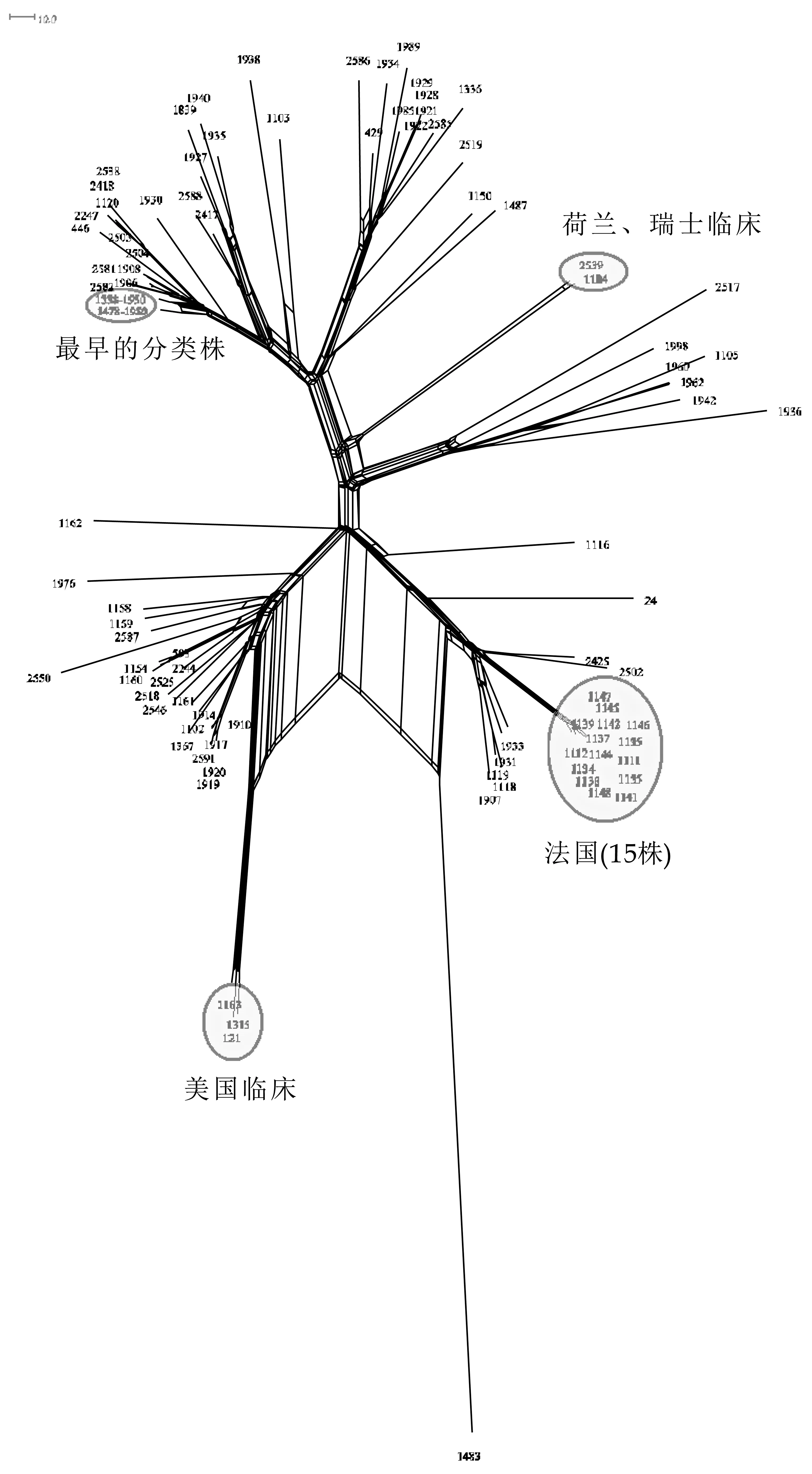
图2 C.sakazakii ST4菌株的cogMLST分析
2.4 C.sakazakii ST13菌株的cogMLST分析
如图3所示,通过Cronobacter Pub MLST数据库中的cog MLST方案对43株阪崎克罗诺杆菌ST 13菌株分析显示,在2005至2006年分离自中国进口乳制品当中的31株菌聚集到了一起,尽管这些乳品进口自6个不同的国家。这一结果与先前Guo等[13]人的SNP分析结果相一致——大多数菌株的SNP位点差异在20以内,因而可推测这些菌株存在克隆关系。值得注意的是,在Guo等[13]人的研究中的菌株CS-71(编号1999)因与参考菌株存在5387个SNP位点差异而与其它31株菌的基因组明显不同。在此次研究当中,菌株CS-71也相对于高度克隆的31株菌形成了一个单独分枝,并且与2016年分离自比利时临床的菌株(编号2505)在基因组层面更为接近。此外,5株在1994年分离自法国临床及婴幼儿配方奶粉中的菌株也形成了一个独立分枝。其余的5株菌则形成了五个不同的分枝。这些结果对于具有相同序列型但来源不同的菌株溯源分析及克隆关系的验证将会有较大帮助。
2.5 C.malonaticus ST 7菌株的cogMLST分析

图3 C.sakazakii ST13菌株的cogMLST分析
如图4所示,35株丙二酸盐阳性克罗诺杆菌ST7菌株的cogMLST聚类分析结果显示,最早在1973年分离自美国临床的菌株(编号71)与1988年分离自新西兰的菌株(编号1485)及2005年分离自韩国的菌株(编号145)基因序列差异较小。两株于2010年分离自中国水环境中的菌株(编号1489和1491)聚集到了一起。此外,两株来源于美国昆虫当中分离时间为2012年的菌株(编号1842和1843)与另外两株源自斯洛文尼亚临床当中分离时间为2017年的菌株(编号2567和2568)分别形成了2个独立分枝。1977年分离自美国临床的两株菌(编号590和1556)也相对于其它菌株形成了一个单独的分枝。在2007年至2013年分离自捷克共和国(编号399和619)和斯洛伐克(编号415)临床的菌株聚集到了一起,另外7株分离自捷克共和国临床的菌株则形成了两个小的分枝。这些结果表明,丙二酸盐阳性克罗诺杆菌ST 7菌株的cogMLST聚类与菌株来源之间存在密切相关性,这对于该序列型致病菌的流行病学调查研究将会有较大帮助。
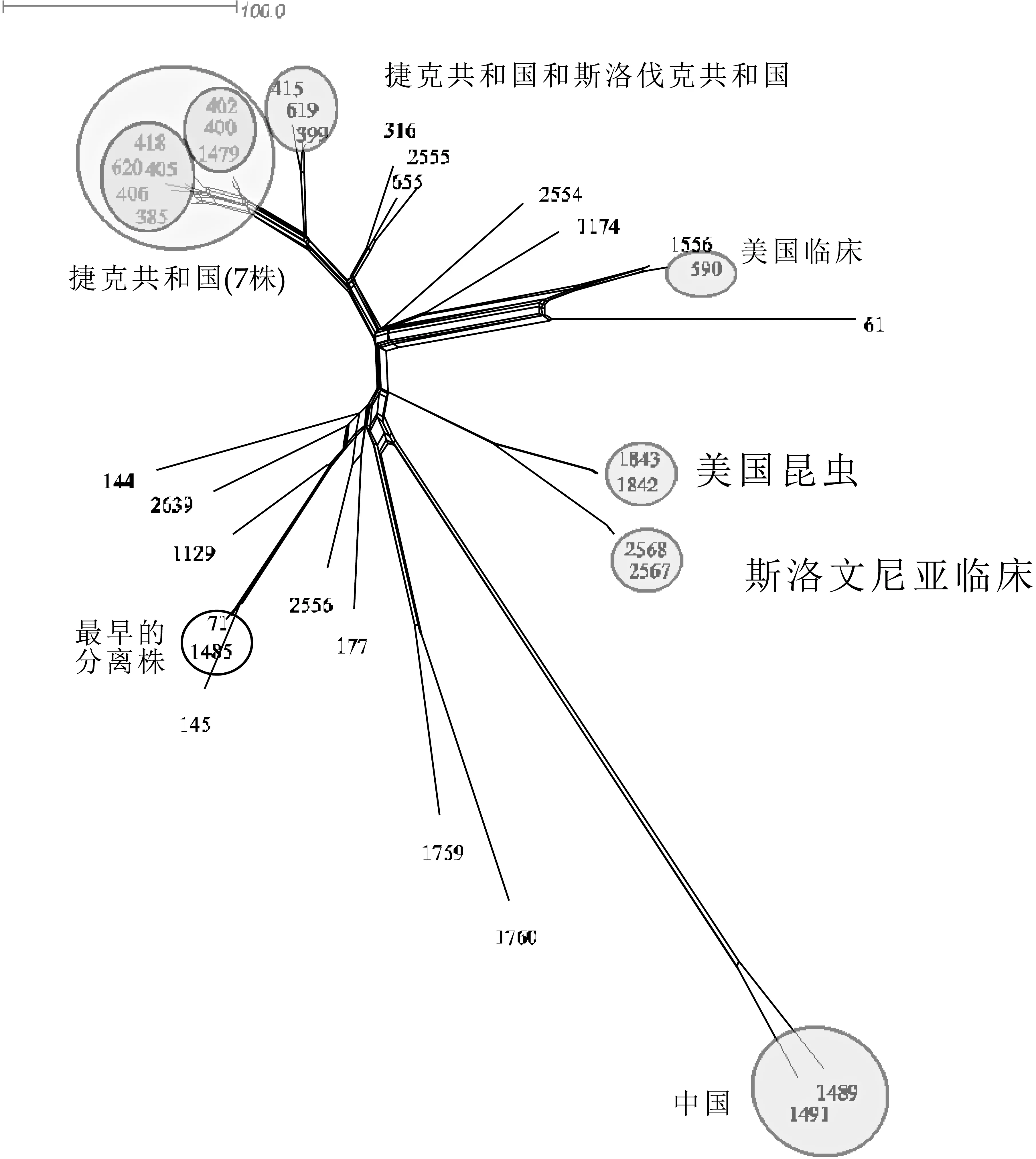
图4 C.malonaticus ST7菌株的cogMLST分析
3 讨论
至今,分类学依旧是关于克罗诺杆菌研究的主题,因为准确的细菌分类对于可靠的监管控制及溯源分析至关重要,而准确的细菌分类则依赖于有效的分型方法[3,9,14]。同时,无论是克罗诺杆菌的流行病学研究还是分子溯源方法的建立都必须基于对目标生物多样性的透彻理解。
对于克罗诺杆菌属各菌株之间的研究,7-loci MLST已被证实是一个非常有用的工具。最初Baldw in等[15]利用7-loci MLST成功地将C.sakazakii和C.malonaticus两个种区分开来,同时结果显示C.sakazakii ST 4菌株为优势菌株(22/60)。2012年Joseph等[16]采用7-loci MLST方法对325株克罗诺杆菌进行系统发育分析进一步证实了7个种的存在。此外,还发现C.sakazakii是临床来源的优势菌种,C.sakazakii ST 4是脑膜炎病例脑脊液分离株的主要序列类型。2014年Forsythe等[9]进一步对C ronobacter PubM LST数据库中的1 007株克罗诺杆菌进行了7-loci MLST分析和统计。结果表明,C.sakazakii克隆复合物(clonal complexes,CC)4菌株与临床来源密切相关,CC 1、CC 4和CC 7为主要序列型。2017年O grodzki等[17]继续采用该方法对1 654株克罗诺杆菌进行了分型和来源分析。结果显示,大多数临床菌株为C.sakazakii CC 1和CC 4,ST 8和ST 12以及C.malonaticus ST 7型。此外,Fei等[18-20]也在近几年持续利用这一方法对分离自中国市售奶粉及乳品厂的克罗诺杆菌污染情况进行了系统的调查分析,不仅揭示了多个主要序列型,而且证明了克罗诺杆菌在中国市售奶粉当中的污染依旧明显。
由于7-loci MLST方法在克诺罗杆菌分型中的有效性,本研究使用7-loci MLST方法对C ronobacter PubM LST数据库中已登记的2 438株菌进行了序列定义及统计分析。7-loci MLST共揭示了591种ST,其中ST1(280株)、ST4(334株)、ST 7(78株)和ST 13(67株)为主要序列型。多数菌株来源于食品及配料当中,并于1950至2017年分离自40多个国家。由于MLST所针对的七个基因位点在基因组总量中占比较小,导致同一ST型中包含地理来源,分离时间,宿主等不相关的菌株,而对菌株溯源结果适得其反。
全基因组测序由于获得的序列信息量足够大,是当前研究克罗诺杆菌分型最有效的方法,并可以对该致病菌进行精确的溯源[21-26]。关于克罗诺杆菌相同ST菌株的溯源分析之前已有研究[13,17,27-28],M asood等[28]采用基于全基因组序列的单核苷酸多态性(Single nucleotide po lym orphism,SNP)分型技术对1994年法国N ICU的C.sakazakii相同ST菌株进行了溯源分析,其结果显示SNP分型方法可以对相同序列型菌株进一步分型,来自不同基因型簇的阪崎肠杆菌分离株都具有感染新生儿的能力,然而爆发来源尚未确定。此外,Guo等[13]也通过利用SNP分析对源自乳品的32株C.sakazakii ST 13菌株进行了分型研究,结果表明,SNP聚簇与原产地及乳制品产品类型之间未呈现有明显的相关性。虽然SNP分析方法具有高度歧视性,但对参考菌株选取较为敏感,即选取不同参考菌株,SNP分型结果存在差异较大[29]。同时SNP分析是计算密集型的,因此分析流程较长;对于分析大型序列集,必须用高性能计算机进行分析。此外,应用该方法进行分析需要大量的生物信息学知识。基于WGS的扩展的多位点序列分型方法[30]相较于SNP分析则具备诸多优势,如其具有稳定的菌株命名法和完善的数据库,能够实现国际标准化和可扩展性,分析速度快、计算要求复杂度低且无需生物信息专业知识。因而本实验并未采用SNP分型技术继续对相同ST菌株进行溯源分析,而是选用了基于全基因组测序的扩展的MLST方法对C ronobacter PubM LST数据库中240株已完成全基因组测序的菌株进行了分型研究。
本研究中所采用的基于全基因组测序的扩展的MLST方法最初是由Forsythe等[9]人建立的cogMLST分析方案,2014年他们利用这一方法对107株克罗诺杆菌进行了全基因组序列分析,进一步证实了C.sakazakii CC 4克隆谱系的稳健性。然而,尚未有报道将该方法应用于相同ST菌株的溯源研究当中。本实验首次利用cog MLST方法对克罗诺杆菌的主要序列型菌株包括:阪崎克罗诺杆菌ST1(67株),ST 4(95株),ST 13(43株)和丙二酸盐阳性克罗诺杆菌ST 7(35株)分别进行了全基因组序列分析。结果显示,该方案可对相同ST内的无关菌株进一步细分,不同聚类与菌株分离国家及来源之间存在一定相关性。
cogMLST分析方法在具有较高流行病学一致性的基础上,能够实现国际标准化,分析速度快,对计算要求复杂度低且无需专业的生物信息知识背景,从而能够快速应用于疾病爆发过程中克罗诺杆菌属菌株的精准溯源分析及高效监管防控当中。此外,通过采用cogMLST方法对克罗诺杆菌的主要序列型菌株的成功细分表明,过去基于单个或多个基因的分型方法由于纳入分析的信息有限,的确误将许多无关菌株划分到了一起。同时,通过结果当中所揭示的不同聚类与菌株分离国家及来源之间的密切相关性,能够进一步表明cogMLST方法对于克罗诺杆菌的溯源分析将是极为有效的分型方法之一。最后,随着越来越多克罗诺杆菌属菌株全基因测序数据的公开,选取有效的全基因组序列分析方法对于相同ST菌株的全球溯源分析及食源性疾病监测至关重要。
