T-RFLP和454焦磷酸测序方法分析制麦过程细菌群落的动态变化
2019-10-29邱然,陆健*
邱 然 ,陆 健 *
(1.江南大学 生物工程学院,江苏 无锡 214122;2.江南大学 工业生物技术教育部重点实验室,江苏 无锡214122;3.粮食发酵工艺与技术国家工程实验室,江南大学,江苏 无锡 214122)
制麦是一个复杂的生物学过程,包括诸多的生理生化反应。大麦在发芽过程中,激活和合成水解酶类,降解蛋白质和淀粉等大分子物质,转化成氨基酸和糖类,供啤酒酵母生长代谢使用[1]。在制麦生态系统中,除了发芽的大麦,还有丰富的微生物菌群[2],其代谢产物对麦芽品质影响很大,并最终影响啤酒质量。微生物的性质和数量会对制麦过程和最终产物啤酒产生有利或不利的影响[3]。因此,深入了解制麦过程中微生物菌落的性质和数量,有利于控制制麦过程和改善麦芽品质,进而提升啤酒质量[4]。
确定大麦和麦芽微生物组成的传统方法是采用涂平板计数和菌落鉴定的方法。这种方法首先要进行微生物培养。在现阶段,自然界中大多数的微生物都是不可培养的,在实验室中能够培养的菌种仅占自然界微生物种类的极小部分,还有相当多的微生物因无法培养而未被人们所认识。因此,传统方法用于研究制麦生态系统中总微生物构成可能会出现严重偏差,导致实验结果与实际不一致[5]。目前,传统方法正逐渐被不需要进行微生物培养的分子生物学方法所补充和替代[6],如指纹识别技术、基因测序等。其中末端限制片段长度多态性(T-RFLP)分析技术不依赖于细菌培养,并且整合了自动测序仪的高分辨率和高通量特征,重现性好,分辨率高,自动化程度高,能迅速产生大量重复、准确的数据。对于分析复杂的群落结构比其他的指纹识别技术具有更明显的优势,已经被广泛用于生态系统和栖息地中的微生物菌群的多样性和分子动力学的研究[7]。随着科技的不断进步,454焦磷酸测序技术已经能快速鉴定微生物菌落,比克隆和Sanger测序更有效,避免了经典分析技术在多样性研究中的局限性以及由此引起的种群丢失、结构不清等弊病,可以直接反映出生态系统中微生物原始的构成情况,也是研究微生物群体多样性的重要手段。然而,到目前为止,还少见T-RFLP和454焦磷酸测序方法用于分析麦芽制造过程中微生物多样性的报道。
乳酸菌在制麦和酿造工业已有较多应用,如利用乳酸菌在制麦和酿造过程中控制腐败和真菌毒素,以提高麦芽质量和安全性[8]。某些特定的乳酸菌会产生抗菌物质与溶解氧竞争,限制有害细菌的生长,缩短麦汁过滤时间[9]。LAITILA等[10]证明在浸麦水中添加乳酸菌可以提高大麦发芽力。尽管在啤酒酿造工业中乳酸菌的重要性已经得到了高度认可,但到目前为止,大部分研究主要集中在个别菌株上[11],与大麦和麦芽制造过程相关的乳酸菌菌群结构和特性尚未得到详细研究。
本研究以两个不同收获年份、同批次但不同制麦方式的大麦为样品,利用T-RFLP技术系统考察了细菌菌落结构变化情况,并重点研究了制麦过程中的乳酸菌。另外,使用454焦磷酸测序技术对乳酸菌16S rRNA基因进行测序,研究制麦过程中内源性乳酸菌菌群特性。
1 材料与方法
1.1 材料和仪器
实验材料:2012年和2013年的国产江苏啤酒大麦单二。麦芽制造由塔式制麦系统和微型制麦系统按照相同的制麦工艺进行。浸麦:4周—7 d—5周—7 d—5周—3 d,浸麦度 43%~44%;发芽:15℃,120 h;干燥:45℃3 h—1 h—55℃3 h—1 h—65℃ 3 h—1 h—70℃ 1 h—1 h—75℃ 1 h—1 h—87℃ 3 h。取样点设在发芽1 d、发芽5 d和最终成品麦芽。所有供试样品由中粮麦芽(大连)有限公司提供。
实验仪器:微型制麦设备,澳大利亚Jeo White麦芽公司产品;PCR仪,Bio-Rad T100,美国伯乐公司产品;变性梯度凝胶电泳仪,Bio-Rad;凝胶成像系统,美国UVP公司;荧光仪,Invitrogen Qubit®2.0,赛默飞世尔公司产品;自动测序仪,373A,美国应用生物系统公司产品。
1.2 细菌总DNA提取
准确称量大麦及麦芽样品150 g装入带有无菌小玻璃珠(212~300 μm)的 500 mL 三角瓶中,向每个瓶中加入 100 mL Tris-HCl缓冲液(pH 8,10 mmol/L),室温,160 r/min 摇床振荡 1 h。离心,弃上清后,将所得沉淀加入液氮在研钵中研磨数次,取出装入1.5 mL离心管中,-20℃冷却保藏,待用。按照文献[4]的方法提取细菌总DNA。
1.3 T-RFLP分析
用于16S rRNA基因T-RFLP分析的2套引物分别为细菌通用引物 516F(5’-TGCCAGCAG CCGCGGTA-3’;5’FAM-labelled) 和 1541R(5’-AAGGAGGTGATCCAGCC-3’);乳酸菌引物 7F(5’-AGAGTTTGATYMTGGCTCAG-3’;5’HEX-labelled)和 677R(5’-CACCGCTACACATGGAG-3’)。其中7F为非特异引物,677R是根据4个重要乳酸菌[12]设计的,包括Lactobacillus、Leuconostoc、Pediococcus和Weissella。
反应体系为20 μL,其中含有0.15 mmol/L的dNTP,0.5 mmol/L引物,1单位的TaqDNA聚合酶,1倍的PCR反应缓冲液,1 μL基因组DNA。
运行程序为:94℃预变性2 min,前30个循环在94℃变性45 s,64℃(通用引物)或66℃(乳酸菌引物)退火45 s,72℃延伸45 s,循环30次,结束后72℃保持10min。被标记的PCR产物(约200 ng)用MspI或HinfI在37℃消化4 h,最后用373A自动测序仪对限制性片段进行分析(重复两次)。
利用T-REX软件分析每个样本的数据和峰值区域。根据电泳图谱对T-RFLP数据进行出峰分类。利用DNA序列限制性酶切位点在线服务平台(http://rna.lundberg.gu.se/cutter2/) 预测 DNA 扩 增片段的酶切位点。对非靶标DNA末端限制性片段去除后,用Sorensen相似性指数分析数据,运用非加权组平均法(UPGMA)进行聚类分析。为了区分峰值与噪音,数据分析仅局限于大于样品总峰值区域1%的TRF。利用微生物菌群分析网站(MiCA)的电脑模拟消化法产生的数据库和本研究的454测序结果来确定微生物种类。MiCA网站有利用核糖体数据库RDP Release 10编辑得到的高质量16S rRNA基因序列。
1.4 16S rRNA基因扩增子的测序
运用454测序仪研究与制麦相关的乳酸菌菌群。用正向引物341F(5’-CCTACGGGAGGCAG CAG-3’)和乳酸菌反向特异引物677R扩增样品。扩增出约423 bp的产物,覆盖了16S rRNA基因V3~V4区域,适合于454焦磷酸测序[13]。对DNA样品进行2次PCR扩增,然后用琼脂糖凝胶电泳对PCR产物进行验证,扩增产物使用Qiagen PCR纯化试剂盒进行纯化。用荧光仪对纯化后的产物定量,取等量浓度产物进行测序。测序过程按照454罗氏GS FLX说明书进行。利用Biopython将序列根据5’和3’特有标记对应到相应的样品。每50 bp移动窗口中,根据Phred最小值25进行序列删减(0.3%的误差率)。最短和最长序列设定为315和330个核苷酸,舍弃不确定的碱基序列和同源多聚体数量多于8的序列。用Uchime嵌合体检测程序检测嵌合体,去除嵌合体后与SILVA参考序列比对。利用临界算法去掉3%的差异序列,将序列分成不同的OTU。通过在GenBank上比对来确定该OTU。再利用核糖体数据库项目(RDP)的网站(http://rdp.cme.msu.edu/)进一步确定结果。将两次乳酸菌数据混合,剔除单体(在所有样本中仅有一个序列的OTU)后,利用Mothur和R生成稀疏曲线,评估样本和OTU数量。
2 结果与讨论
2.1T-RFLP分析
为了研究制麦过程中微生物菌群结构,采用TRFLP技术研究完整的细菌菌群和乳酸菌菌群。末端限制性片段长度多态性方法是将PCR引物中的一条进行荧光标记,用合适的限制性内切酶将PCR扩增产物消化,在酶切后产生的许多不同长度的限制性片段中只有末端带有荧光标记的片段才能被监测到。通过分析这些荧光信号生成的T-RFLP图谱,可以揭示样品中微生物的数量、种类和种群规模,从而帮助了解微生物菌群的结构、功能及动态变化。
2.1.1 细菌菌群 在14个样本中(见图1),MspI消化获得38个不同细菌的TRF,HinfI消化获得53个TRF。每个样品用MspI消化后的TRF数量为4~16个,用HinfI消化后的TRF数量为6~26个,说明制麦过程细菌多样性相对有限。图1为采用TRFLP 技术分析收获于 2012 年[图 1(a)]和 2013 年[图1(b)]江苏单二大麦在不同制麦方式下微生物末端限制片段的相对数量和分布情况(HinfI消化,取两次实验的平均值。峰面积超过所有样品总面积的10%的TRF被保留,其他 TRF列为一组“其他TRFs”)。几乎所有样品都有少数的特征TRF。所有样本中有6个TRF总量超过3%,包括136、332、630、738、796 bp和824 bp,分别占总样本的4%、3%、7%、16%、18%和20%。样本中均有136、738 bp和824 bp的TRF。796 bp的TRF存在所有非大麦样品中。这些TRF对应种属如下:136 bp:Enterobacter和Sphingobacterium;332 bp:Wautersiella和Cryseobacterium;630 bp:Curtobacterium和Propionobacterium;738 bp:Weissella,Lactobacillus,Lactococcus和Streptococcus;796 bp:Acinetobacter和Stenotrophomonas;824 bp:Leuconostoc和Pseudomonas。

图1 T-RFLP分析制麦微生物末端限制片段的相对数量和分布情况Fig.1 Relative abundance and distribution of the most dominant bacterial terminal restriction fragments obtained by T-RFLP analysis
其 中Lactobacillus、Lactococcus、Streptococcus和Weissella常见于发芽过程[14]。大麦中的TRF个数约占所有样品的75%,也就是说制麦环境中增加了1/3的外源微生物。
为了比较不同样品中细菌菌落结构的区别,用NTsys 2.1e软件对T-RFLP数据进行聚类分析。聚类结果表明,样品基本可以根据工艺步骤进行分组(图1(a)),说明细菌菌落结构随着制麦工序的不同而变化。两个年份大麦样品被分为一组;发芽过程的绿麦芽样品和最终的麦芽样品分为一组。不同发芽方式之间并没有明显的区别。另外,相同年份的绿麦芽和最终成品麦芽相似度更高。大麦和麦芽的细菌菌落相似性只有12%。微生物菌群结构的变化主要是因为大麦在发芽过程所处环境不同所引起,如发芽温度16~20℃,非常适合微生物生长繁殖,导致微生物菌落在整个发芽过程显著增加。同样,成品麦芽经过80~85℃的高温焙焦,杀灭了大部分绿麦芽上的微生物,导致成品麦芽中微生物数量和种类减少。

图2 非加权组平均法(UPGMA)聚类图Fig.2 Unweighted-pair group method with arithmetic mean(UPGMA) dendrogram
2.1.2 乳酸菌菌群 经过T-RFLP分析,乳酸菌有30个不同的TRF,用MspI酶切后每个样品有4~15个TRF。用乳酸菌特异性引物(7F-677R)检测乳酸菌Lactobacillus、Leuconostoc、Pediococcus和Weissella,与整个细菌菌落相似,主要的TRF存在于所有研究样品中,如图3所示。然而,与整个细菌菌落相反,在过程阶段未进行区分,其树形图相对较分散,见图2(b)。T-RFLP分析是基于扩增片段经过酶切后长度不同。所有样品中3个最主要的TRF分别对应553、578 bp和583 bp,相应峰面积分别为39%、15%和23%。根据这些TRF的模拟酶切结果,推测这 3个 TRF分别是Leuconostoc、Lactobacillus和Weissella。

图3 T-RFLP分析制麦乳酸菌末端限制片段的相对数量和分布情况Fig.3 Relative abundance and distribution of the most dominant bacterial terminal restriction fragments representing lactic acid bacteria obtained by TRFLP analysis
2.2 乳酸菌16S rRNA基因扩增子的焦磷酸测序
采用454焦磷酸测序技术重点研究乳酸菌菌群情况,排除3%以上差异的序列后分成139个运算分类单元(OUT),其中102个与乳酸菌有关。发芽大麦和麦芽的非乳酸菌序列占比很少,约2%。大麦的非乳酸菌序列占比较多,约13%,这可能是由于大麦样品的乳酸菌含量较低。传统MRS平板计数实验表明,大麦中细菌有107cfu/g,乳酸菌大约有1×103cfu/g,发芽后直至麦芽中乳酸菌含量达到1×106~1×108cfu/g[15]。所以大麦样品中存在较多的非乳酸菌序列可能是因为引物错配导致一些非特异DNA被扩增。除去单体后,与乳酸菌相关的序列有33624个,平均317 bp,属于47个OTU。每个样本序列数量为517~4874(平均 2402),每个 OUT 序列数量为2~9548。有14个OTU序列数量大于所有样品总数的0.1%,这14个主要的OTU占每个样本中乳酸菌序列超过98%,如表1和图4所示。在所有样本中,至少有97%的序列与大麦样本有关,表明在麦芽生产过程中,大麦携带的微生物起着重要作用,T-RFLP分析也证明了此结论。此外,5个主要的 OUT:OTU100、101、102 为Weissella属(魏斯菌属),OTU093为Lactobacillus属(乳杆菌属)和OTU098为Leuconostoc属(明串珠菌属)。这5个OUT在样品中均存在,覆盖率达88%。
魏斯菌属是制麦过程中最主要的乳酸菌,有3个主要OTU,占所有序列的63%。乳杆菌属是第2优势属,占所有序列的20%。第3是明串珠菌属,占所有序列的11%。由图4可知,2个不同年份的样品间乳酸菌菌群存在巨大差异:2012年的乳杆菌比2013年更丰富,2013年收获的大麦乳杆菌相对含量与2012年相比急剧下降;魏斯菌属在2013年更加丰富,在整个过程中相对含量始终较高,平均为82%。不同年份魏斯菌属的主要OUT也不同,2012年以OTU102为主,而2013年以OTU101为主。其次,两种发芽方式主要的种属也不同:2012年塔式制麦以乳酸杆菌为主,而微型制麦以魏斯菌属为主。此外,Pediococcus几乎只在2012年的微型制麦系统中检测到,发芽1 d后占乳酸菌菌群11.2%,5 d后占4.6%,占麦芽的12.3%。对于大麦来说,片球菌在2012年占32.6%,在2013年含量较低,仅占大麦乳酸菌菌群1.6%,在制麦过程中约占0.1%。

表1 制麦过程乳酸菌主要运算分类单元Table1 Lactic acid bacteria operational taxonomic units identified in this study
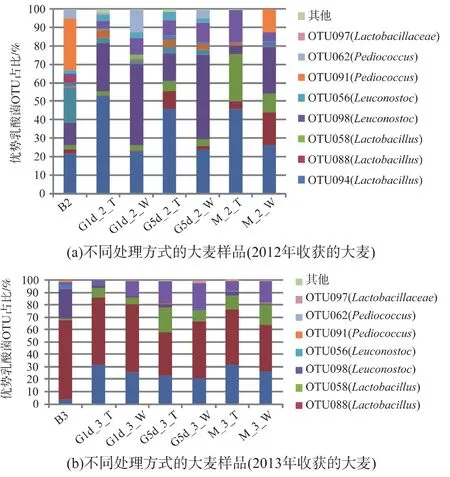
图4 采用454焦磷酸测序技术分析制麦过程中优势乳酸菌的相对数量和分布情况Fig.4 Relative abundance and distribution of the most dominantlacticacid bacteria during malting based on 454 pyrosequencing
与T-RFLP相比,聚类分析基本是把样品按照年份分组,见图2(c)。用454焦磷酸测序分析发现:在麦芽生产过程中,不同制麦方式的同一批次大麦中乳酸菌菌落结构具有显著差异。例如,Pediococcus仅在微型制麦系统中被检测到。据报道,Pediococcus可以提高麦芽质量[10]:有利于制麦过程中有益酵母生长并且限制有害细菌和镰刀真菌生长,同时提高麦芽的特性,包括降低麦汁粘度和β-葡聚糖含量,提高木聚糖酶和微生物β-葡聚糖酶活性,最终改善麦芽过滤性能。另外,微型制麦的麦芽脆度较高,未溶解的大麦较少,这些都有助于提高麦芽质量。但Pediococcus是否起作用还有待进一步研究。
3 结 语
在制麦过程中,大麦中整个细菌菌落组成有限,通过T-RFLP分析发现菌落结构在制麦过程中是变化的,可能是在制麦过程中受环境影响所致。454焦磷酸测序法分析乳酸菌菌群结果表明:在样品中均有 5个 OTU,分别是代表了Weissella、Lactobacillus和Leuconostoc。454测序结果还表明,不同年份收获的大麦其菌群结构不同,而T-RFLP法并未检测到这点。此外,454测序结果显示:同一批次大麦在不同的制麦系统中乳酸菌菌落结构也存在显著不同。
T-RFLP和454焦磷酸测序结果都证明大麦本身携带的微生物种类与制麦过程条件无关。然而,微生物的相对数量却由制麦过程工艺参数决定。随着对这些微生物的深入了解及有效控制,本研究结果将作为微生物制麦技术的基础依据。
