环氧乙烷灭菌产品的无菌放行
2019-10-19刘文一李道成徐星岗
刘文一 李道成 徐星岗
1 嘉兴高是灭菌技术有限公司 (浙江 嘉兴 314005)
2 武汉致众科技股份有限公司 (湖北 武汉 430040)
3 苏州市倍咏医疗科技有限公司 (江苏 苏州 215104)
内容提要: 依据《中华人民共和国医疗器械监管法规》《中华人民共和国药典》、国家标准GB 14233.2以及GB 18279.1,探讨医疗器械环氧乙烷灭菌后的几种无菌放行程序,分析这几种无菌放行程序的关系以及优缺点,同时总结国际上欧美等发达国家环氧乙烷灭菌产品常用的无菌放行程序。按照GB 18279.1的传统放行或者参数放行可以避免无菌检查法的局限性,确保无菌的要求得到完全满足,提高产品流通效率,避免资源的浪费。
环氧乙烷灭菌后的无菌医疗器械在制造商加工完成后,需要进行无菌放行。国内比较通行的无菌放行方式为按照《中华人民共和国药典》(简称《中国药典》)的无菌检查法进行无菌检测,但是该方法用于常规灭菌放行存在很多局限性。而新版GB18279.1对于环氧乙烷灭菌后的产品规定了另外的无菌放行的方法,避免了无菌检查法的局限,其等同采用的ISO 11135-1也是目前国际上医疗器械发达国家通用的无菌放行法。目前,国内医疗器械监管机构和企业在这方面的认识还不统一,依据无菌检查法放行无菌医疗器械不仅没有科学的依据,也不能真正起到监管的作用。为此本文研究了产品放行的法律法规要求,对比了药典无菌检查法和专用灭菌法的无菌放行法之间的优缺点,并详细的介绍了GB 18279.1-2015标准给出的环氧乙烷灭菌产品的放行程序。
1.无菌医疗器械的法规介绍
无菌医疗器械是一种特殊类型的医疗器械,通常情况下,无菌医疗器械是接触人体或者侵入人体的,因为人体在接触到外来的细菌、病毒时容易引发患者感染,因此产品需要达到无菌的要求。所谓无菌,是指产品上无存活微生物的状态,理论上讲是一种绝对的情况,然而这种理论上的绝对概念与统计学的检验是有矛盾的。因此工业实践中,定义了无菌保证水平(SAL)来确定无菌的具体量化要求,其无菌保证水平(SAL)应不超过10-6,是无菌医疗器械必须要达到的确保其能安全和有效的关键性能。无菌性能的质量监控也就是判定整批产品的阳性概率是制造商必须履行的无菌放行程序。

表1. 批出厂产品最少量检验数量
无菌放行目前已知的方法有如下几种:①按照《中国药典》2015版,第四部1101无菌检查法进行,其中关于医疗器具的批出厂产品最少量检验数量的要求,见表1。②按照GB 14233.2-2005[1]进行,但由于这份标准直接引用了《中国药典》,因此实际就是药典的无菌检查法。③按照GB 18279.1-2015,10常规监视和控制以及11产品灭菌放行的规定进行。
由于按照GB 14233.2-2005进行的无菌检查法实际上就是按照《中国药典》无菌检查法进行的,因此以上3种放行的方法实际上是2种无菌放行方法。即无菌检查法放行以及非无菌检查法放行。
2.药典无菌检查法的局限性
无菌检查法用于无菌产品常规放行是存在很大局限性的,制定者非常明白无菌检查法存在着无法规避的风险,因此,在应用《中国药典》[2]或者GB 14233.2标准[1]时都做了相应的申明“若供试品符合无菌检查法的规定,仅表明供试品在该检验条件下未发现微生物污染”“相关产品标准在引用本部分时应注意根据产品应用特性规定适宜的合格判定指标”。无菌检查法存在的缺陷如下。
2.1 接收质量限(AQL)和过无菌保证水平(SAL)的矛盾
环氧乙烷灭菌过程是特殊过程,过程复杂,流程长,参数多,灭菌的关键参数受到设备、环境、装载、产品、包装等因素的影响,实际的灭菌过程中,各个参数波动也比较大,而无菌检查法依赖于抽样,对于灭菌批而言,抽样抽到灭菌最差的情况实际是不可能的,而均匀抽样存在一个可信度的问题,GB 2828系列标准是抽样的依据标准,在确定抽样方案时候,需要确定一个AQL(接收质量限),无菌保证水平SAL不大于10-6也就是接收质量限AQL应达到10-6。无法在GB 2828标准中找到合适的抽样方案。
而根据《中国药典》的抽样方案,如表1,从中取1例查下GB 2828标准的合格质量限AQL。
例1:某批环氧乙烷灭菌的注射器,其批量是2万支,按照药典的抽样方案,应抽取20件。为了简化评估,采用了正常抽样检验一次抽样方案,查表会发现样本量20件对应的接收质量限是0.65,这个质量限度远远不能保证SAL,整整相差0.65%/10-6=65000倍,而实际上按照药典的任何一个抽样方案,其接收质量限都远远不能保证SAL[3]。
因此,根本上说,无菌检查法无法满足无菌医疗器械的最基本性能要求。
2.2 无菌检查法存在的较大的误判风险
例1中,可以查GB 2828标准,得知正常检验的生产方风险,是12.0,表示不接受0.65的质量限的灭菌批的概率是12.0%,也就是制造商采用这个无菌检查法的抽样方案,即使合格保证水平才0.65%,仍然有12%的概率会误判产品是不合格的[3]。
在一批产品中,检出污染品的概率(P)与取样量存在如下关系,见公式(1)[4]。

式中,P代表了检出污染品的概率;Nu代表了灭菌之后的污染概率;n代表检查的阳性数。上述算式可转化为公式(2):

计算示例如下:
例2:某批产品实际的污染率为千分之一(Nu=1.0×10-3),无菌检查抽样数为1000,无菌检查结果呈阳性的概率是(P),则:
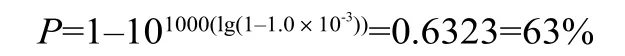
例3:某批产品实际的污染率为百万分之一(Nu=1.0×10-6),要保证95%的阳性检出率,计算抽样量n,则:
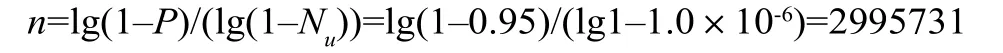
案例3表明,需要测试约3×106个样品,发现只有一个样品出现阳性,或全部阴性,才能有95%的把握证实实际污染率符合无菌保证水平(SAL≤10-6)[4]。
根据公式(2),可以计算得出表2无菌检查发现阳性结果的概率(P)、样品数(n)和实际污染率(Nu)之间的对应关系。
表2数据表明,通过无菌检查法发现污染品的概率(P)取决于该批产品的实际污染率以及随机抽样数(n)。当污染率为10-3时,样本量低于100,实际检出阳性的概率低于10%,当污染率为10-6时,样本量低于10000,实际检出阳性的概率低于1%[4]。
在实际的无菌检查实验中,因人的因素或者是技术条件等导致无菌检查呈现的假阳性概率大约在0.1%~20%[5,6]。陈万胜[7]采用了直接接种法,薄膜过滤法制作了400个样本,观察两种样本制作方法菌培养假阳性率,结果显示,直接接种法假阳性率8.5%,薄膜过滤法假阳性率4%,总假阳性率为6.25%。
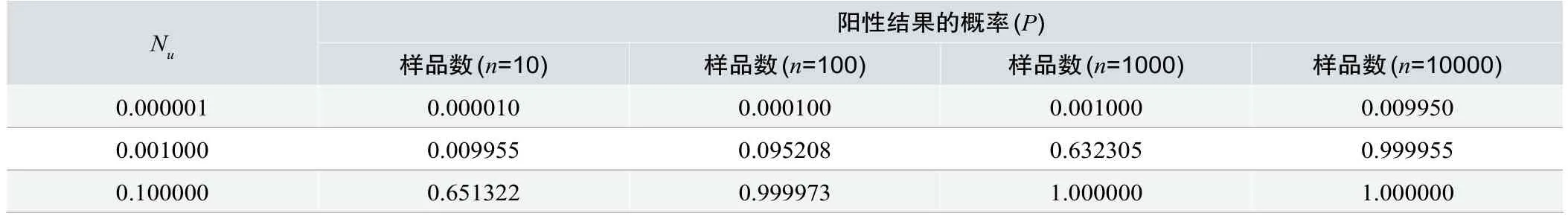
表2. 阳性结果的概率(P)、样品数(n)和实际污染率(Nu)之间的对应关系
显然通过无菌检查法来证实一批产品的无菌保证水平是不切实际的。
2.3 产品无菌培养时间长的缺陷
产品无菌培养,需要14d,培养时间长可能使培养基性质改变以及损害微生物复苏。另外,到目前为止也没有单一的培养基能培养出所有微生物,因此,实际的无菌检查法往往需要进行细菌和真菌两项测试,甚至更多培养基的测试[8]。此外,14d的培养时间还需要保存至少14d库存,以及占用巨大的库房,给制造企业带来了极大的不便。
2.4 复杂的产品结构带来的检测技术缺陷
越来越多的大型医疗器械,譬如医疗手术包的无菌检查,很难找到合适的浸提器皿,器皿越大,越容易污染,因此,需要采用SIP,这又带来了一系列的风险,如样品分割时的污染、SIP能否代表整个产品等。
2.5 破坏性检测带来的巨大成本缺陷
有很多无菌医疗器械成本非常昂贵,如心脏起搏器、带药支架等,其材料、加工等成本均价格不菲,而按照《中国药典》的产品无菌检查法进行测试,最少的样本量也需要4件,无菌检查大部分是破坏性试验,每次测试浪费的财力甚是可观。
2.6 典型样本选择非常困难
环氧乙烷灭菌确认时最难灭菌样品需要考虑非常多的因素,如装载结构、灭菌器结构、温度最低点、产品结构等因素,如采用无菌检查法,也应该采用灭菌确认中选择出的最难灭菌的样本进行无菌实验,但实际的常规灭菌采用这样的样本几乎不可能。
3.GB18279的无菌放行法的优点
研究发现,微生物在灭菌剂的作用下,影响环氧乙烷灭菌效果的一些关键参数稳定时,单位时间内死亡的微生物数量比例是相对稳定的。如图1所示[9]。
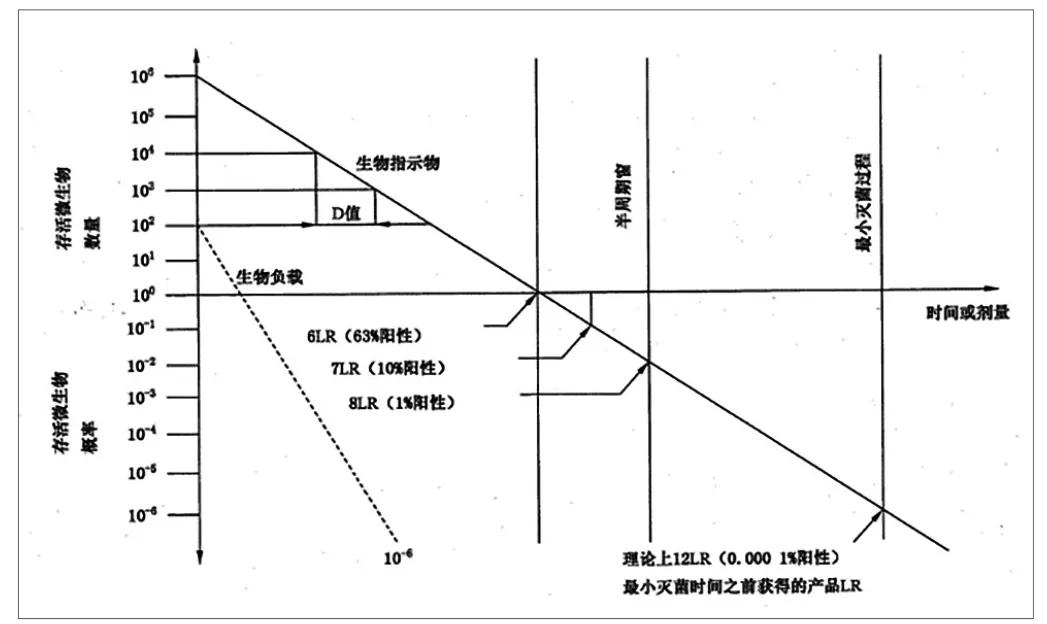
图1. 生物指示剂和生物负载的死亡曲线
在图1中,生物指示剂在环氧乙烷作用下,其存活的微生物数量(概率)的Log值是正比于时间的。在6D时,此时微生物的存活概率是63%,12D时,此时微生物的存活概率是10-6,正是研究需要的SAL。图1中的黑实线是生物指示剂的死亡曲线,而在医疗器械的制造过程中,对于产品加工的环境均有严格的要求,产品初始污染菌(生物负载)的抗性绝大部分是不如BI的特殊菌株的,数量也是大大低于BI(106)的,因此,只要常规环氧乙烷灭菌的参数符合确认的参数规定,其合格保证书平就可以远远低于上述的SAL的要求,如上图虚线所示。工业实践中,灭菌器参数的保证还通过常规控制、设备维护保养、变更控制、年度评审、再确认等管理要求保证其可靠性。
因此,按照GB 18279.1标准进行环氧乙烷灭菌后产品的无菌放行,其优势在于:①可靠性高,SAL完全得到了满足,甚至可以做到过度杀灭。②误判的风险降低:灭菌器参数可靠性非常高,尤其最新的灭菌器的设计,温度、湿度、压力等传感器均会采用双传感器进行监控,甚至很多企业开始使用浓度传感器直接监测灭菌器内的环氧乙烷浓度,大大降低了灭菌失败的可能,另外,BI的无菌检测技术相对产品的无菌检测技术,由于BI很小,培养基是优化过的,BI无菌测试操作更简单,因此失败的风险也比较低。③无菌放行成本低廉,无需破坏大型的复杂的产品,也无需大型的浸提器皿,甚至自含式的BI根本无需超净的实验室。④无菌放行的时间短,如采用参数放行,灭菌结束即可放行(实际还得考虑解析时间),如采用BI无菌检测+参数放行,则BI无菌检测需要7d(实际还得考虑解析时间),自含式的BI可以缩短到2d。⑤EPCD的放置非常简单,BI无菌检查的取样非常容易。
4.GB18279的无菌放行法的实际应用
在欧美等医疗器械发达国家,按照环氧乙烷专用灭菌标准给定的无菌放行法早已普遍应用。而我国,早在2005年,国家食品药品监督管理局就已经批准无锡华瑞制药有限公司和广州百特医疗用品有限公司进行了参数放行的试点[4]。另外,输液器标准GB 8368对无菌放行的规定,描述也是“输液器宜按GB18278、GB18279或GB18280对灭菌过程进行确认和进行常规控制,以保证产品上的存活概率小于10-6”。
下面介绍GB 18279.1[10]给出的两种无菌放行方法。
4.1 传统放行
传统放行是2000年颁布的GB 18279提出的概念,2015年颁布的GB 18279.1删除了此概念,但是实际的放行条件还是和2000年颁布的GB 18279的传统放行一样,仍可以称为“传统放行”。
具体的做法是记录并保存每一个灭菌批的灭菌周期数据,并对这些数据进行审核,应完全符合经确认的灭菌过程规范,灭菌过程规范应按照GB18279.1标准规定建立,并经过过程确认。
同时还应按照确认的灭菌过程规范进行BI的无菌检测,BI应全部阴性,阳性对照应阳性。
上述要求,全部满足可以判定产品无菌。
4.2 参数放行
参数放行是一种不使用生物指示剂来放行经灭菌的无菌产品的方法,而依靠证明物理处理参数均符合所有的规范来放行无菌产品,在上述传统放行灭菌周期数据的基础上增加如下的三项参数记录要求:①在整个灭菌周期,至少从2个位置记录灭菌温度数据;②在处理阶段(加湿和保湿),直接测定柜内的湿度;③按照规定的时间间隔,通过直接分析灭菌柜内的气体确定环氧乙烷浓度。
5.无菌检查法的实际应用
无菌检查法虽然不能用于制造商常规的灭菌产品放行,但是有几种情况,还必须用到无菌检查法。
5.1 市场抽检
这不是医疗器械制造商的判定,抽检是政府行为,本身就是对市场上的产品进行监督检查,无法也不能从制造商获得灭菌参数以及BI的培养结果来判定产品是否无菌,另外,市场上的产品是否无菌还和产品的包装、运输等相关,因此必须按照药典的要求进行无菌检查。
5.2 灭菌确认过程中的抗性比对
灭菌确认过程中,需要评价产品本身的抗性和IPCD(过程挑战器械)的抗性,必须进行短周期下的产品无菌测试,此时,应按照无菌检查法进行。
5.3 周期性的性能测试
为了定期评估产品全性能,通常对产品留样应进行年度的全性能测试,此时应进行产品无菌测试,当然需要按照无菌检查法进行。
6.小结
对于无菌医疗器械制造商而言,采用环氧乙烷灭菌后,按照药典的无菌检查法进行产品的无菌放行,存在很多的缺陷,例如SAL水平得不到满足、较高的误判风险、较长的检测时间、复杂的检测技术、较高的检查成本等,应当采用GB 18279.1中规定的产品无菌放行方法进行,可以确保完全满足SAL的要求,是完全符合《医疗器械生产企业质量控制与成品放行指南》[11]规定的“确保每批成品都符合接收准则”的要求的。
事实上,不仅仅是环氧乙烷灭菌的产品有特定的无菌放行方法,目前已经建立的专用的灭菌标准中,都有相应的灭菌放行方法,如蒸汽灭菌可以按照GB 18278.1[12],辐射灭菌可以按照GB 18280.1规定的灭菌后的产品放行程序进行。
因此,对于无菌医疗器械的制造商,在制定企业的技术规范时,应该参照相应的专用灭菌标准规定无菌放行的方法。目前,很多制造商的技术规范在已经备案的情况下,可以按照《医疗器械生产企业质量控制与成品放行指南》[11]的规定“给出经过确认的替代解决方案。”从而在企业内部建立无菌放行的程序,以替代技术规范中用无菌检查法放行产品的描述,同时积极的变更技术规范。
