SUZ12过表达及敲减恶性外周神经鞘瘤稳定细胞株的建立及其意义
2019-10-17李新宇朱香熹孙硕遥刘嘉岳赵玉龙李光明
张 静 ,高 雅 ,李新宇 ,朱香熹 ,孙硕遥 ,肖 可 ,王 静 ,刘嘉岳 ,赵玉龙 ,李光明 ,朱 泽
(1.天津医科大学病原生物学系,天津300070;2.天津医科大学基础医学院,天津300070;3.遵义医科大学珠海校区临床医学系,珠海519090;4.多伦多大学圣乔治校区,多伦多ONM5S)
恶性外周神经鞘瘤(malignant peripheral nerve sheath tumor,MPNST)是一类具有高度恶性,发展迅速,易转移复发且预后差的侵袭性软组织肉瘤,约占所有软组织肉瘤的5%~10%,其中将近50%的MPNST与NF1基因相关,NF1的病人一生中有10%~15%的风险转变成MPNST[1-4]。另外,肿瘤抑制因子(TP53和p16INK4A)的丧失和生长因子受体信号(例如EGFR或PDGFR)过度激活在神经纤维瘤的恶性转化中也起着重要作用。因此,基于这种关联,MPNST最经典基因学研究大多集中在NF1基因的缺失以及TP53和CDKN2A/p16基因的缺失上[5]。然而最近的一些对MPNST病人的组织标本进行二代测序的研究结果显示,在MPNST中PRC2的组成单元SUZ12和EDD的突变失活也属频发事件[5-9]。PRC2可以调节组蛋白H3的27位点上的酪氨酸三甲基化,而SUZ12作为PRC2复合体的重要组分,可以通过稳定PRC2复合体保证H3K27me3在表观遗传修饰过程中功能正常发挥。
CRISPR/Cas9是在细菌和古细菌中发现的一种获得性免疫系统,通过crRNA识别、Cas蛋白裂解来抵抗病毒和噬菌体的入侵,因其敲除效率高,脱靶率低,敲除效果稳定的优点被用于替代传统的TALEN技术和ZEN技术,已成为一种主流的基因编辑工具[10-11]。因此,为了进一步研究SUZ12基因在MPNST中的作用,笔者使用慢病毒载体构建SUZ12过表达及CRISPR/Cas9敲减的MPNST稳定细胞株,为针对SUZ12在MPNST中的生物学功能、作用机制和疾病诊断治疗的进一步研究提供了实验基础。
1 材料与方法
1.1 主要材料和试剂 ST88-14细胞株由天津医科大学肿瘤医院杨吉龙教授馈赠;293T细胞和大肠杆菌由本实验保存;胎牛血清、DMEM培养基购自美国Gibco公司;慢病毒载体系统(GV492、GV371、pHelper1.0、pHelper2.0),Lenti-CAS9-puro 慢病毒购自上海吉凯公司;质粒抽提试剂盒购自Promega公司;TOP10感受态细胞购自Genechem公司;FastQuant RT Kit、SuperRealPreMix Plus购自天根公司;SUZ12引物由北京索真公司合成;MTT试剂盒购自索莱宝公司。
1.2 细胞培养 MPNST细胞系ST88-14在含10%的胎牛血清和1%的双抗的DMEM完全培养基中培养,37℃,7.5% CO2温箱中孵育。
1.3 SUZ12基因过表达载体的构建
1.3.1 SUZ12目的基因PCR扩增 SUZ12基因PCR扩增引物,正向引物:5′-AGGTCGACTCTAGA GGATCCCGCCACCATGGCGCCTCAGAAGCACGGC GGTG-3′,反向引物:5′-TCCTTGTAGTCCATACCG AGTTTTTGTTTTTTGCTCTGTTTTG-3′,扩增产物为2 261bp。PCR 体系反应条件:98℃ 5 min,98℃ 10 s,55 ℃ 10 s,72 ℃ 90 s,循环 30次,72 ℃ 8 min。扩增完成后,1.5%琼脂糖凝胶电泳测定结果。
1.3.2 过表达载体的构建 将GV492载体用BamHI/AgeI酶切,PCR产物与载体进行交换,构建重组质粒(设置一组不含SUZ12片段的质粒作为对照组)。重组质粒转化入感受态细胞,将其涂布在含有氨苄青霉素的平板上,在37℃培养箱中培养过夜,之后进行菌落PCR的鉴定,正义链:5′-GATGTTAATGAAGGAGAGAAAG-3′,反义链:5′-CCTTATAGTCCTTATCATCGTC-3′。产物经1.5%琼脂糖凝胶电泳,选取阳性克隆进行转化,挑菌,质粒抽提,进行测序鉴定。
1.4 SUZ12敲减载体的构建
1.4.1 sgRNA靶位点的设计与合成 在NCBI中明确SUZ12基因的CDS外显子区域,使用麻省理工学院的CRISPR设计工具(http//crispr.mit.edu/)设计3对SUZ12基因的sgRNA序列,并在其两端加入BbsⅠ位点,序列见表1。引物退火形成带有粘性末端的双链,稀释200倍后使用。

表1 3对特异性sgRNA序列Tab 1 3 pairs of specific sgRNA sequences
1.4.2 sgRNA载体的构建 将GV371载体用BbsⅠ酶切,与退火形成双链的sgRNA混合,配置酶切连接反应体系进行连接,酶切体系:Vector plasmid(100 ng/μL)1 μL,Oligo 双链 DNA(0.089 μmol/L)2 μL,10×Buffer Tango 2 μL,DTT(10 mmol/L)1 μL,ATP (10 mmol/L)1 μL,T7 DNA Ligase 0.5 μL,BbsI 1 μL,H2O 11.5 μL;将上述反应物置于 PCR 仪,37 ℃5 min,21 ℃ 5 min,16 ℃ 30 min,共 6 个循环。将连接产物转化至TOP10感受态细胞中,接种到含Amp抗性的LB固体培养基上,37℃培养箱中过夜。挑菌,质粒抽提,进行测序鉴定。
1.5 慢病毒包装与滴度测定
1.5.1 慢病毒包装 将GV载体质粒、pHelper1.0载体质粒、pHelper 2.0载体质粒按照一定比例(20 μg∶15 μg∶10 μg) 加到 2.4 mL 的 Opti-MEM 和 100 μL的Lipofectamine2000中,共同转染293T细胞,37℃、5% CO2培养箱中培养。48 h后收集293T细胞上清液,超速离心去除上清,加入病毒保存液,充分溶解后,高速离心,分装上清,-80℃保存。
1.5.2 慢病毒的滴度检测 测定前24 h,接种293T细胞到96孔板(4×104个/孔):在EP管中加入90 μL的无血清培养基,取待测的病毒原液10 μL加入到EP管中,倍比稀释到最后一管;弃去293T细胞培养基,加入90 μL稀释好的病毒溶液,37℃培养箱培养;24 h后加入完全培养基,96 h后观察绿色荧光蛋白的表达情况。
1.6 确定慢病毒感染的最适MOI,以及嘌呤霉素(Puro)筛选的最适剂量
1.6.1 感染预实验 ST88-14细胞在病毒感染前一天铺板,至细胞汇合度为20%~30%;病毒于冰上融化,依次将病毒稀释至滴度 1×108TU/mL,1×107TU/mL,1×106TU/mL,吸弃培养液加入病毒及相应感染增强液,混匀,继续培养,12 h后换回常规培养基,继续培养;感染72 h后,用荧光显微镜观察感染效率。
1.6.2 确定嘌呤霉素的最适剂量 将ST88-14细胞接种于48孔板中,48 h后加入Puro进行筛选,Puro 浓度梯度设置为 0、0.5、1、2、4、6 μg/mL;Puro处理48 h后细胞全部死亡的最低药物浓度作为筛选稳定株的药物浓度。
1.7 慢病毒感染ST88-14细胞及稳定细胞株的建立1.7.1 过表达稳定株的构建 制备密度为3~5×104个/mL的ST88-14细胞悬液,每孔100 μL接种到96孔板中,37℃培养24 h;病毒冰上融化,根据预实验MOI值进行稀释;吸弃培养液加入病毒及相应感染增强液,在37℃、7.5% CO2温箱中培养,12h后换回常规培养基,继续培养;感染72 h后,加入2 μg/mL的Puro筛选48 h以上。之后降低Puro浓度到1 μg/mL,继续对细胞进行筛选和扩增,同时收集细胞行下游RT-qPCR检测。
1.7.2 CRISPR/Cas9敲除稳定株的构建 制备密度为 3~5×104个/mL 的细胞悬液,每孔 100 μL 接种到96孔板中,37℃培养24 h;Cas9病毒冰上融化,根据预实验MOI值适当稀释;吸弃培养液加入病毒及相应感染增强液,在37℃,7.5% CO2温箱中培养,12 h后换回常规培养基,继续培养;感染72 h后,加入2 μg/μL的 puromycin筛选至少 48 h以上,得到ST88-14-Cas9细胞。将ST88-14-Cas9细胞铺6孔板,用含1 μg/mL剂量的Puro的完全培养基培养至30%,按照预实验感染条件分别感染3个sgRNA的Lenti-sgRNA-GFP病毒,12 h后换回常规培养液,感染48~72 h后,在荧光显微镜下观察绿色荧光的表达情况。用含1 μg/mL的Puro的完全培养基扩大培养,继续传代、扩增进行鉴定。
1.8 SUZ12过表达和敲减稳定株的鉴定
1.8.1 荧光显微镜观察 用含1 μg/mL Puro筛选两周后,用倒置荧光显微镜观察绿色荧光蛋白的表达效率,并拍照。
1.8.2 RT-qPCR鉴定 用TRIzol提取SUZ12过表达组和敲减组总RNA,反转录得到cDNA,进行RT-qPCR 检测,SUZ12 引物序列为:SUZ12-F:AGAAAACGAAATCGTGAGGATGG;SUZ12-R:GCAC GTAGGTCCCTGAGAAA。PCR反应体系为:cDNA template 4 μL,Primer F (20 μmol/L)2 μL,Primer R(20 μmol/L)2 μL,SYBR qPCR Master Mix 10 μL,Rox Dye 0.04 μL,加水至 20 μL;PCR 反应条件:98℃ 2 min,98℃ 10 s,68 ℃ 30 s,循环 40次,95 ℃ 15 s,60℃1 min,99℃15 s。每组实验独立重复3次。
1.9 SUZ12过表达和敲减组细胞增殖活性的测定 胰酶消化各组细胞,调整细胞浓度为1×104个/mL,每孔200 μL接种到96孔板;分别在 24、48、72 h后弃上清,加入90 μL完全培养基和10 μL MTT溶液,孵箱内继续培养4 h;弃上清,每孔加入110 μL DMSO溶液,避光震荡10 min,490 nm处测吸光度,绘制增长曲线。每组实验独立重复3次。
1.10 统计学处理 采用SPSS20.0处理数据,多组间均数比较采用方差分析。P<0.05有统计学意义。
2 结果
2.1 过表达重组质粒的鉴定结果 过表达重组质粒SUZ12的PCR产物经琼脂糖凝胶电泳检测后得到一个2 261bp的特异性条带,与预期大小一致,见图1。GV492载体经BamHI/AgeI酶切后与SUZ12基因构建重组质粒,转化、涂板、挑菌后进行PCR鉴定,经琼脂糖凝胶电泳分析,得到469bp的阳性转化子,见图2。酶切鉴定阳性的克隆经过测序,结果与NCBI中的人SUZ12基因(ID:23512)进行比对,序列一致,表明过表达重组载体构建成功。

图1 SUZ12基因扩增电泳图Fig 1 PCR amplification of SUZ12

图2 重组载体PCR产物鉴定及琼脂糖凝胶电泳图Fig 2 Identification of recombinant vector and agarose gel electrophoresis
2.2 CRISPR/Cas9表达载体的构建 为了提高敲除效率,设计并合成了3条sgRNA靶向序列,载体GV371(U6-sgRNA-SV40-EGFP)BbsⅠ酶切后,分别与退火获得的3个目的片段sgRNA连接过夜,转化TOP10感受态细胞、涂板、挑菌、质粒抽提、送测序鉴定。测序结果表明插入的片段序列与设计合成的靶向SUZ12序列一致,见图3。

图3 sgRNA表达载体阳性克隆转化子测序峰图Fig 3 sgRNAexpressionvectorpositiveclonetransformantsequencing peak map
2.3 病毒滴度的测定 在倒置荧光显微镜下观察加入不同稀释倍数病毒原液的293T细胞荧光表达情况显示,荧光细胞的数目随病毒稀释倍数的增加而逐渐减少,估算过表达慢病毒和3种Lenti-sgRNA的病毒滴度分别为 5×108TU/mL、1×109TU/mL、1×109TU/mL 和 1.5×109TU/mL,见图 4。

图4 不同稀释倍数的慢病毒感染293T细胞的荧光表达情况(×200)Fig 4 Fluorescenceexpressionsof293Tcellsinfectedwithlentiviruses at different dilutions(×200)
2.4 慢病毒感染的最适MOI及嘌呤霉素筛选的最适剂量 在不同浓度病毒感染ST88-14细胞72 h后,用显微镜观察荧光表达丰度,感染效率80%左右,细胞生长良好的组所对应的MOI值即可作为后续感染实验的依据,确定MOI=10为CRISPR/Cas9慢病毒的最佳感染条件,MOI=100为过表达慢病毒的最佳感染条件,见图5。待ST88-14细胞生长至70%~80%时加入不同浓度的Puro,筛选48 h以上,当Puro的浓度大于等于2 μg/mL时,ST88-14细胞全部死亡,因而确定Puro最适剂量为2 μg/mL,见图6。
2.5 慢病毒转染ST88-14细胞的结果 慢病毒转染ST88-14细胞2周后,用倒置荧光显微镜进行观察,镜下均可见大量绿色荧光蛋白,并且荧光随着细胞传代可持续表达(图7)。
2.6 稳定株细胞的筛选及鉴定 构建好的SUZ12过表达及敲减慢病毒分别感染ST88-14细胞后,经Puro筛选获得稳定细胞株,提取RNA进行RT-qPCR检测。PT-qPCR结果显示过表达组SUZ12 mRNA表达量明显较对照组高(P<0.05),敲减组SUZ12 mRNA表达量相比对照组明显减少(P<0.05)。由此说明,SUZ12过表达和敲减的ST88-14稳定细胞株构建成功,见图8。

图5 过表达组相同感染条件下不同浓度病毒感染效率比较(×200)Fig 5 Comparison of virus infection efficiency at different concentrations in the same infection conditions(×200)

图6 不同浓度嘌呤霉素筛选48 h后ST88-14的细胞状态Fig 6 Cell state of ST88-14 after 48 hours at different concentrations of puromycin

图7 慢病毒转染ST88-14细胞后稳定表达绿色荧光蛋白(×200)Fig 7 Stable expression of green fluorescent protein after transfection of ST88-14 cells with lentivirus(×200)

图8 过表达和敲减组SUZ12 mRNA相对表达情况Fig 8 Relative expression of SUZ12 mRNA in overexpressed and knockdown groups
2.7 SUZ12基因过表达抑制MPNST细胞增殖 SUZ12过表达和敲减稳定株构建完成后,用MTT法测定各组细胞增殖活性的改变。MTT结果显示,过表达组的细胞增殖活性与阴性对照组相比明显降低(P<0.05),敲减组的细胞增殖活性与阴性对照组相比明显升高(P<0.05)。表明SUZ12基因过表达抑制MPNST细胞增殖。见图9。
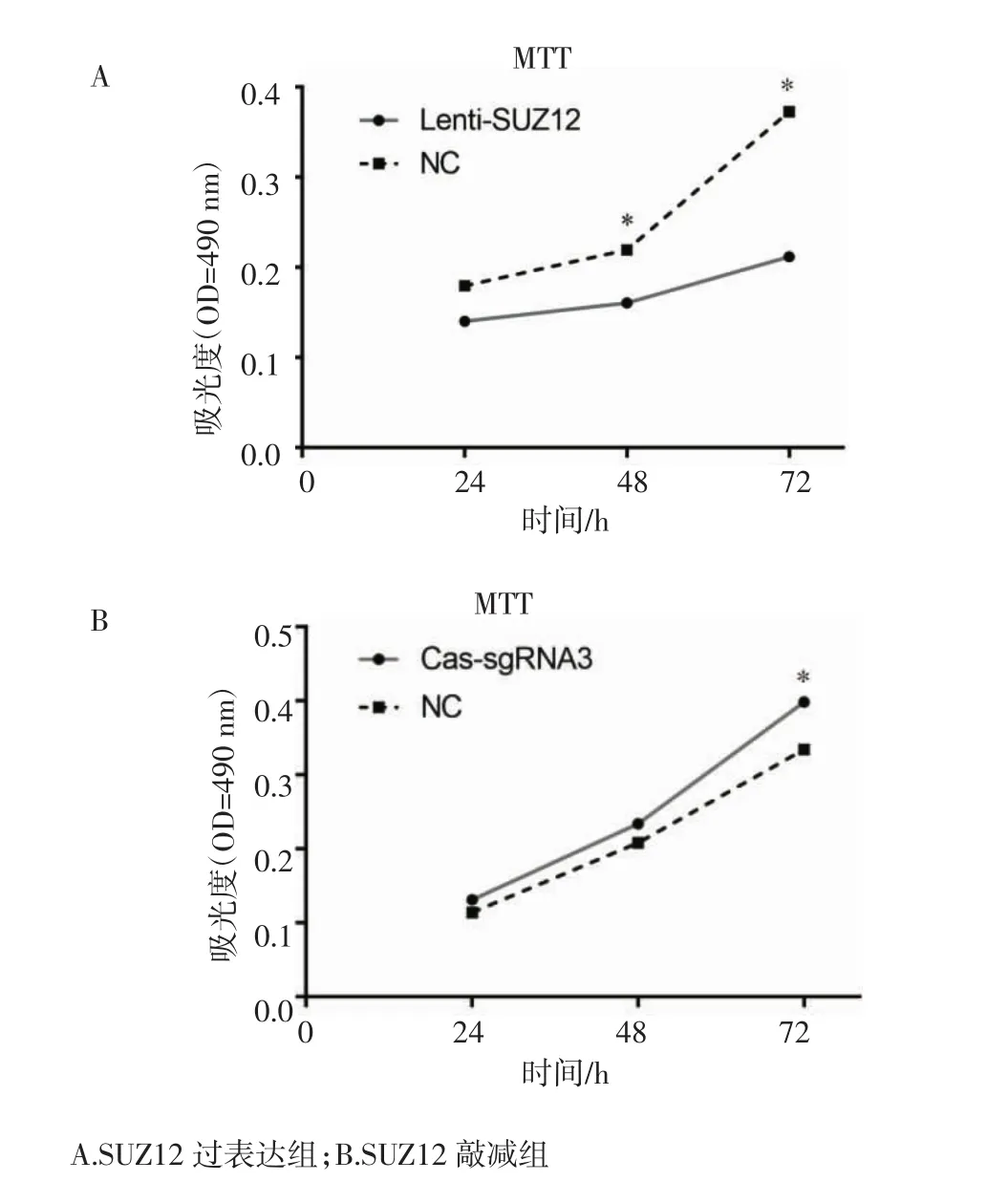
图9 过表达和敲除组细胞增殖活性的测定Fig 9 Determination of cell proliferation activity in overexpression and knockout groups
3 讨论
SUZ12是多疏抑制复合物2(PRC2)必不可少的组成部分,它可以通过调控组蛋白和DNA甲基化修饰转录过程,借此以稳定PRC2,使PRC2的功能正常发挥[12],除此之外,SUZ12还可以影响组蛋白三甲基化的活性,参与EED-EZH2复合物的功能沉默过程[13]。有研究表明SUZ12在胃癌、结肠癌、卵巢癌、子宫内膜癌、乳腺癌、套淋巴细胞癌等肿瘤中表达明显高于癌旁组织,并且,SUZ12表达量的增加提高了肿瘤细胞无限增殖、转移及侵袭的能力,影响了患者的预后[14-18]。SUZ12在上述肿瘤中表达量增加,促进了肿瘤的发展,然而其在血液系统肿瘤、神经胶质瘤等肿瘤中的表达量明显低于癌旁组织,这表明SUZ12表达量的减少也促进了肿瘤的发展,同时具有抑癌的作用[19-20],因而,SUZ12在肿瘤的发生发展过程中发挥了双向功能。
本研究组在对12例MPNST全基因组进行深度测序后发现,在NF1阴性的散发性MPNST中,SUZ12基因往往呈现缺失状态[3],这提示我们SUZ12可能在散发性MPNST的发生发展过程中起到了关键作用。Andrew等结合了全外显子测序队列的分析、TCGA提供的公开数据,以及对该疾病的前几代测序研究的回顾,分析计算出了MPNST特异性基因突变率,分别为 NF1(56/64=87.5%),SUZ12(69/123=56.1%),EED(40/123=32.5%),TP53(29/72=40.3%),CDKN2A(54/72=75.0%)[5-9],也证实在 MPNST 中大多存在SUZ12基因的缺失。研究发现大多数MPNST患者中存在PRC2不同组分功能丧失性突变,而SUZ12和EED的缺失性突变又阻断了H3K27me3的甲基化进程,导致H3K27me3缺失,引起PRC2染色质调节信号通路失活[21]。研究者在10%~25%的肾癌、肺癌、胃癌和神经胶质瘤中发现了H3K36me3的缺失[22-25],而H3K36me3的缺失与细胞周期检查点相关基因存在协同致死效应,WEE1抑制剂AZD1775对H3K36me3缺失的细胞更加敏感[26],本课题组之前对43例MPNST的组织标本进行免疫组化染色,发现H3K27me3在65.11%的MPNST患者中均缺失[27],而H3K36me3缺失可以作为用药靶点也为由PRC2组分突变所致H3K27me3缺失的肿瘤提供了一个表观遗传学的治疗策略。
本课题组所用的肿瘤细胞系属于神经细胞,而神经细胞具有不可分裂的特性,使外源片段的导入比较困难,慢病毒载体是由人类免疫缺陷病毒(HIV-1)改造后产生,具有感染效率高、基因片段容量大、持续稳定表达且可感染分裂和非分裂细胞的特点,因此选用慢病毒载体作为基因操作的工具。CRISPR/Cas9是一项新兴的基因编辑技术,自问世以来,已广泛应用于肿瘤领域的科学研究,而且有研究表明,CRISPR/Cas9即使不进行单克隆筛选,也可永久显著敲低目的基因的表达水平[28]。本实验通过慢病毒包装系统制备了SUZ12过表达和CRISPR/Cas9敲减慢病毒,分别转染ST88-14细胞后,成功获得了能稳定表达绿色荧光蛋白的SUZ12过表达和敲减稳定细胞株,RT-qPCR的结果也证明过表达组SUZ12 mRNA表达量明显较对照组高(P<0.05),敲减组SUZ12 mRNA表达量相比对照组明显减少(P<0.05)。MTT结果显示,过表达组的细胞增殖活性与阴性对照组相比明显降低(P<0.05),敲减组的细胞增殖活性与阴性对照组相比明显升高(P<0.05)。由此说明,SUZ12基因过表达抑制MPNST细胞增殖。
综上所述,笔者通过本课题组和其他实验室的研究数据确定了SUZ12基因在MPNST中处于缺失状态,而H3K36me3缺失可以作为治疗靶点也为PRC2组分缺失突变所致的H3K27me3缺失提供了治疗策略,因此笔者构建了SUZ12过表达和敲减的MPNST稳定细胞株,初步探索SUZ12基因表达状态的变化所引起细胞增殖的改变,本课题组将进一步在体内和体外实验中探索细胞侵袭迁移、周期等功能的改变,以阐明PRC2功能失活在MPNST的发病演进中的作用及其分子机制,并为MPNST的治疗提供了一个基于表观遗传学的治疗策略。
