基于高通量测序对中国不同区域传统发酵香肠细菌多样性的研究
2019-10-10黄郑朝宋莲军黄现青乔明武赵秋艳张平安刘茜
黄郑朝,宋莲军*,黄现青,乔明武,赵秋艳,张平安,刘茜
1(河南农业大学 食品科学技术学院,河南 郑州,450002) 2(郑州市大豆深加工重点实验室,河南 郑州,450002)
发酵香肠是指将绞碎的肉、脂肪、糖、食盐、发酵剂、香辛料等混合,再灌进肠衣,经过微生物发酵而制成的具有典型发酵风味的肉制品[1-2]。因其具有风味独特[3-4]、营养价值高[5]、储藏时间长等特点,深受全世界消费者的喜爱。发酵香肠根据含水量、产地、发酵程度和加工时间有不同的分类标准而分成不同的产品[6]。中式香肠通常采用自然发酵方式来制作香肠,由于其微生物复杂多样,香肠的安全和品质没有质量保障。中式香肠相较于国外工业化生产的香肠来说,拥有自己稳定和独特的微生物群落,并且不同地区发酵香肠的微生物群落之间可能具有一定差异,而不同微生物对香肠的品质有极大的影响,因此研究各个地区发酵香肠微生物分布显得尤为重要。
微生物发酵剂具有可以在发酵过程中产酸和促进水分活度(Aw)下降的能力,可以抑制香肠腐败和致病微生物的生长,从而延长产品的货架期[7]。中式香肠作为传统肉制品最为典型的代表,其生产方式较为落后,不仅加工期长,产品感官欠佳,还存在亚硝胺、生物胺和有害微生物等隐患[8],菌种筛选在提高香肠食用安全方面一直是研究重点,研究人员在菌种复配和降低香肠生物胺含量方面做了大量研究[9-13]。当前大多数对发酵剂的研究都是在传统微生物技术的基础上,从环境中筛选符合预期的菌种,并不能把环境中所有的微生物进行分离、纯化和培养,也不能说明各菌种之间的相互交叉作用,这显然对研究结果有一定影响。近年来,随着高通量测序技术的发展,为难培养微生物的研究提供了技术支持,和传统微生物技术相比,该方法可以快速获取样品中所有微生物的信息并且已广泛应用到食品、医学和环境科学等领域[14-15]。
本文采用高通量454焦磷酸测序技术,克服了传统微生物技术耗时耗力和研究不够全面的缺陷,可以避免细菌在挑菌落不全面、没有真正显示出微生物类群的漏洞。收集自中国4个不同地区的发酵香肠,可以在一定程度上反映中式香肠细菌群落的分布情况,为中式香肠的安全性研究和下一步香肠发酵剂的筛选提供一定的理论支撑。
1 材料与方法
1.1 试验材料
2018年3月于四川成都(SC)、广东深圳(GD)、湖南长沙(HN)和黑龙江哈尔滨(HB)地区分别购买传统发酵香肠,每个地区购买5种,采集的样品均来自当地菜市场或超市,购买时样品均已在常温下储藏1个月左右,并采用真空包装的形式在常温条件下运输,样品采集完成后立即进行后续实验。
一步式细菌DNAout试剂盒:北京天恩泽基因科技有限公司;聚合酶链式反应(polymerase chain reaction,PCR)用酶:宝生物工程(大连)有限公司。
1.2 仪器与设备
DYY-6C型电泳仪,北京市六一仪器厂;PCR仪,北京东胜创新生物科技有限公司;凝胶成像分析系统,上海勤翔科学仪器有限公司;SX-50型高压蒸汽灭菌锅,北京五洲东方科技有限发展公司;Neofuge-1600R型冷冻高速离心机,上海力申科学仪器有限公司;1300型生物安全柜,赛默飞世尔科技公司;BagMixer40型拍击式均质机,Interscience公司;DH-420电热恒温培养箱,上海树立仪器仪表有限公司。
1.3 试验方法
1.3.1 试验分组
香肠根据不同的地区分为5组, SC香肠为A组,5种分别编号为A1,A2,A3,A4和A5; GD香肠为B组,5个种类分别编号为B1,B2,B3,B4和B5;HN香肠为C组,5个种类编号分别为C1,C2,C3,C4和C5;HB香肠为D组,5个种类分别编号为D1,D2,D3,D4和D5。
1.3.2 PCR扩增细菌16S rDNA
在无菌条件下,香肠去肠衣,随机取样25 g,装入无菌均质袋中,加入150 g无菌生理盐水,拍击式均质器拍打(2 min)提取细菌,取1.5 mL提取液,离心去除上清液留沉淀备用,按照天恩泽一步式细菌DNAout试剂盒使用说明书提取细菌DNA。338F(5′-ACTCCTACGGGAGGCAGCA-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)作为扩增细菌16S rRNA基因V3-V4区域的引物。PCR扩增总体积为20 μL,其中含有4 μL 5×FastPfu缓冲液,0.8 μL正向引物(5 μmol/L),0.8 μL反向引物(5 μmol/L),2 μL 5 mmol/L dNTPs,0.4 μL FastPfu聚合酶,0.2 μL BSA,10 ng模板DNA,加ddH2O至20 μL。PCR参数设置参数如下:95 ℃下变性3 min;循环数为36次, 95 ℃下变性30 s,50 ℃下退火30 s,72 ℃下延伸45 s; 72 ℃下延伸10 min。3 μL PCR产物用2%琼脂糖凝胶电泳进行质检。
1.3.3 文库构建和上机测序
使用TruSeq® DNA PCR-Free Sample Preparation Kit建库试剂盒进行文库构建,构建好的文库经过Qubit和实时荧光定量PCR,文库合格后,使用v2测序试剂盒(2×250 bp)和MiSeq测序仪进行上机测序[16]。
1.3.4 数据处理
每个样品进行3次重复试验,所有实验数据均用SPSS 11.0软件进行数据处理,采用单因素方差分析进行多样本均数比较,检验水准为α=0.05,采用Origin 8.0软件作图。
2 结果与分析
2.1 Pan和Core OTU分析
OTU(operational taxonomic units)是在系统发生学或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,属,种、分组等)设置的统一标志[17]。Pan OTU指的是泛OTU,是所有样本所包含的OTU的总和,用于观测随着样本总数的增加,总OTU数目的增加情况。Core OTU指的是核心OTU,是所有样本所共有的OTU总数目,用于观测随着样本总数的增加,共有OTU数目的减少情况。从图1可以看出,随着样品数量的增加,曲线的变化近似于渐近线,说明样本量已经足够反映客观事实。

A-Pan OTU分析;B-Core OTU分析图1 Pan和Core OTU分析Fig.1 Pan and Core OTU analysis
2.2 细菌多样性分析
2.2.1 细菌Alpha多样性分析
Alpha多样性是指一个特定区域或者生态系统内的多样性,常用的度量标准有Sobs、Chao1、Shannon和Simpson指数。Sobs是用来表示丰富度实际观测值的指标,Chao1指数是生态学中用来估计物种总数的常用指数。Shannon指数越大,说明群落多样性越高;Simpson指数越小,说明群落多样性和均匀性越高[18]。4个不同地区之间发酵香肠细菌多样性指数如表1所示。在四川地区中,A5组的Sobs(160.33±2.08)、Shannon(2.10±0.25)和Chao1(197.47±2.32)指数要明显大于其他4组(P<0.05),Simpson指数要明显小于其他4组(P<0.05),A5组在SC香肠中细菌多样性最丰富。在广东地区中,B3组的Sobs(252.00±4.00)、Chao1(266.79±4.32)和Shannon(3.22±0.06)指数要明显大于其他4组(P<0.05),B3组Simpson(0.11±0.01)指数要明显小于其他4组(P<0.05),B3组在GD香肠中细菌多样性最丰富。在湖南地区中,C4组的Sobs(208.66±21.38)指数要明显大于其他4组(P<0.05);Chao1(246.81±25.81)指数要明显大于其他4组(P<0.05),其中C4和C1组之间不存在显著性差异。C1组的Simpson指数要明显小于其他4组(P<0.05),其中C1组和C2组之间不存在显著性差异;C1组的Shannon指数要明显大于其他4组(P<0.05),C4组在HN香肠中细菌多样性最丰富。在哈尔滨地区中,D3组的Sobs指数要明显大于其他4组(P<0.05);Shannon指数要明显大于其他4组(P<0.05),其中D3组和D5组之间不存在显著性差异;Simpson指数要明显小于其他4组(P<0.05),其中D3组和D5组之间不存在显著性差异;Chao1指数也要明显大于其他4组(P<0.05),D3组在HB香肠中细菌多样性最丰富。

表1 细菌多样性指数表Table1 Bacterial diversity index
注:相同地区之间不同字母表示具有显著性差异(P<0.05)。
2.2.2 细菌Beta多样性分析
PCOA(principal coordinate analysis)是将聚类分析与主成分分析方法结合起来,用较少的主坐标对分类单元进行有效的排序,不同分组样本的距离越近,表明样本物种组成越相近,从图2中可以看出,哈尔滨香肠和其他3个地区之间有明显的不同,广东和湖南香肠之间没有明显的区分,两者的细菌在OTU水平上的分布基本上接近一致,四川香肠里面有少部分样品在OTU水平上的分布同广东和湖南相似,剩余样品同其他3个地区之间有明显的不同。
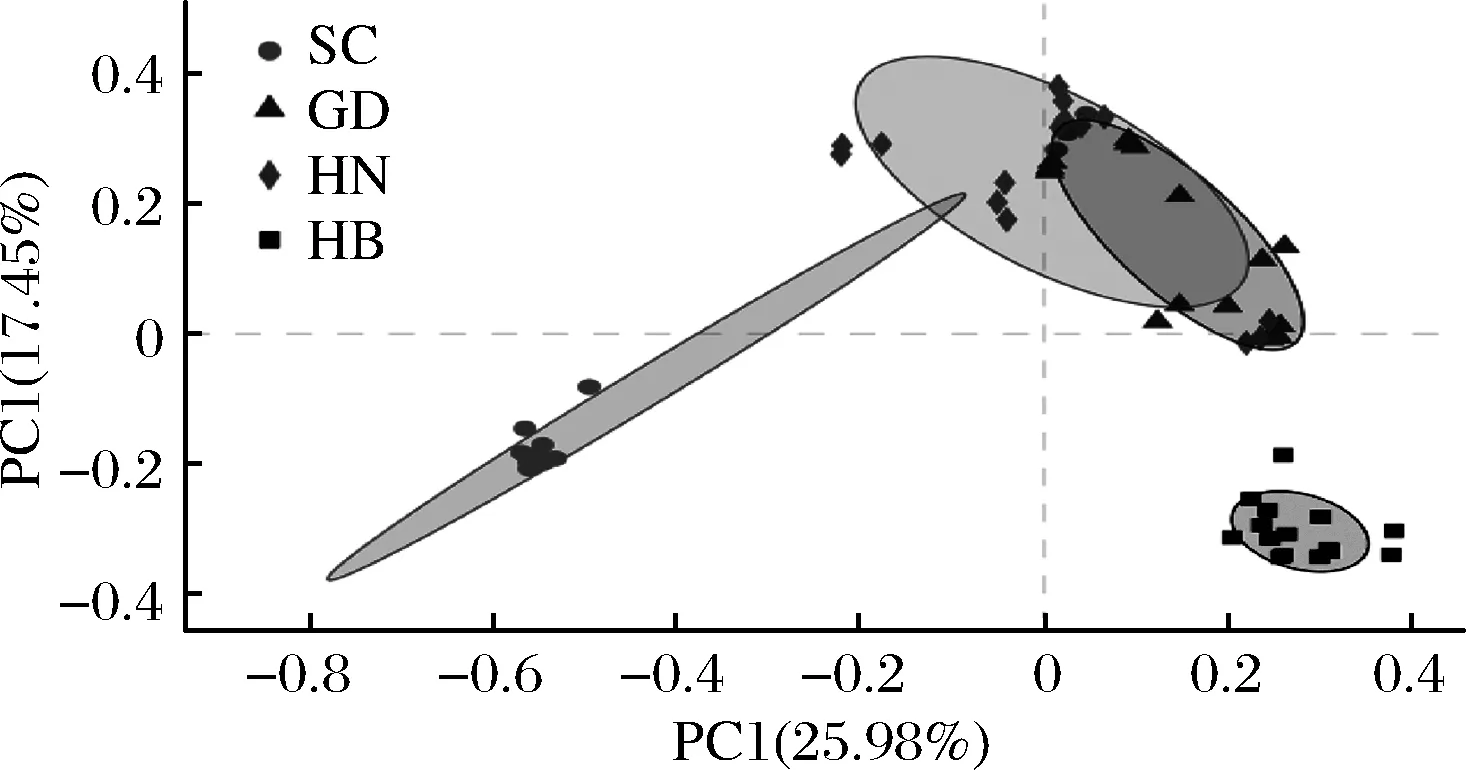
图2 细菌Bray-Curtis距离的PCOA图Fig.2 PCOA plot of bacterial Bray-Curtis distance
2.3 细菌组成分析
图3是对各个地区不同样品的有效序列进行聚类之后求均值,细菌在菌门水平上的分布图(相对含量<1%归为others)。4个地区60个样品共检测到6个菌门,其中厚壁菌门、蓝藻门和变形菌门在所有地区的样本中占多数,是样品中的优势菌门,但是三者在不同区域样品中的相对丰度不一样,在SC和HN中,厚壁菌门占据绝对优势,在GD中,三者的相对含量大致相同,而在HB中,蓝藻门是绝对的优势菌门。
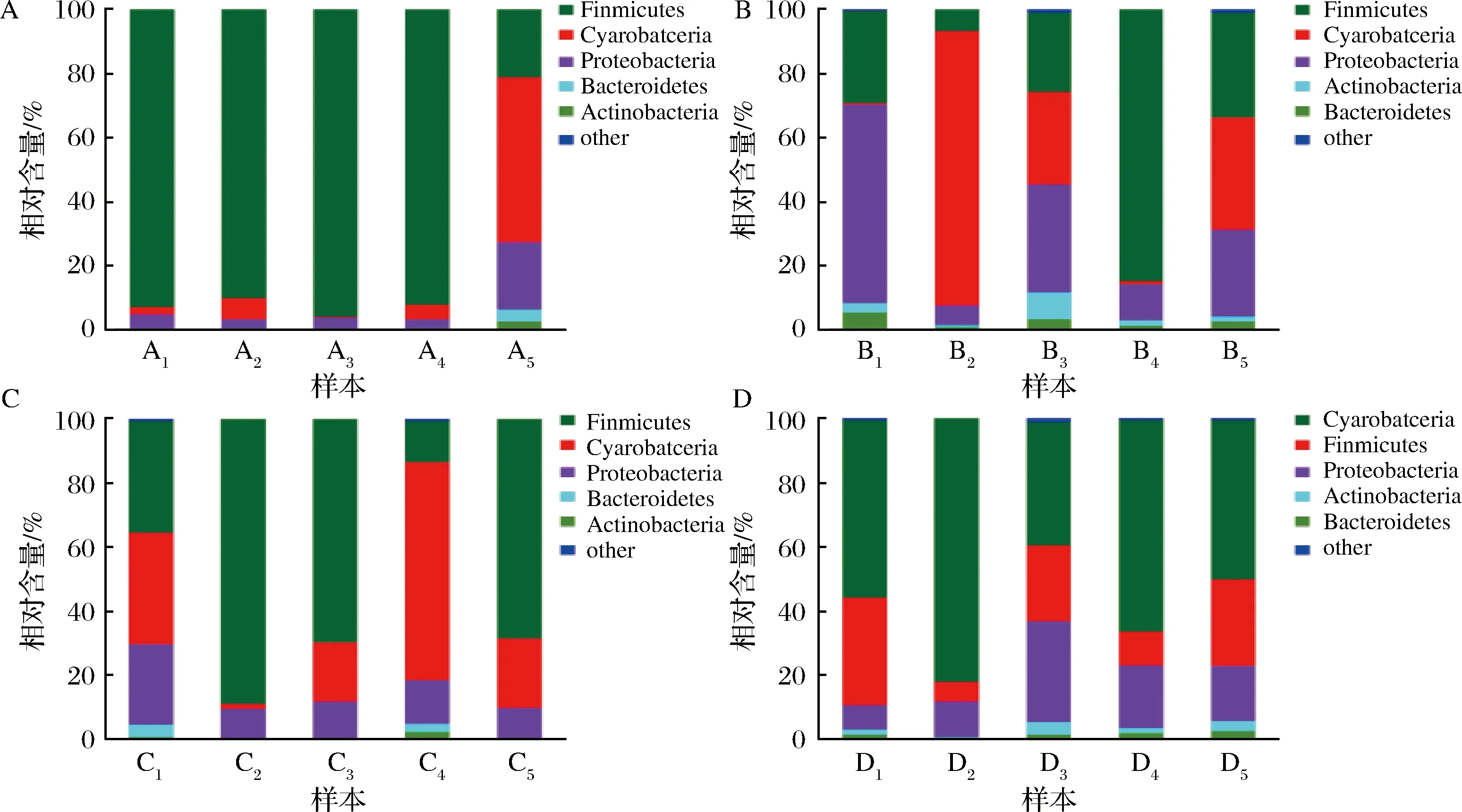
A-SC 发酵香肠; B-GD发酵香肠; C-HN 发酵香肠; D-HB发酵香肠图3 细菌群落结构门水平分布图Fig.3 Bacterial community structure phylum level distribution map
图4是对各个地区不同样品的有效序列进行聚类之后求均值,细菌在菌属水平上的分布图(相对含量<1%归为others),表2是不同地区优势菌属的分布表(占比前3)。所有样品中共检测到26个菌属,4个地区中均含有无法比对的菌属存在。其中乳杆菌属是SC的绝对优势菌属,相对含量为53.68%,乳球菌属在GD和HN中的相对含量较高,分别为17.62%和16.04%。在HB中,并没有哪一种菌属存在较大的优势,所有的菌属分布较为均匀,反而无法比对的菌属占据了绝大优势,相对含量达到了58.13%,对这些无法确定的细菌还需进一步研究;巨型球菌属是除了在HB地区之外都检测到的优势菌属。
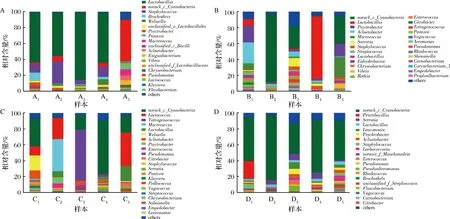
A-SC发酵香肠 B-GD 发酵香肠; C-HN 发酵香肠; D-HB 发酵香肠图4 细菌群落结构属水平分布图Fig.4 Distribution of bacterial community structure genus level distribution map

表2 不同地区优势菌分布表Table 2 Distribution of dominant bacteria in different regions
3 讨论
本研究采用高通量测序技术对4个地区共20种香肠中的细菌群落进行分析,结果发现,从细菌多样性和OTU重叠角度分析,HN和GD香肠的细菌组成比其他地区要更加丰富;从主坐标角度分析,SC和HB香肠更具有地区性。哈尔滨香肠是烟熏熟制而成,并且湖南长沙和广东深圳两者都属于亚热带季风气候,而四川成都和哈尔滨分别属于亚热带季风性湿润气候和温带季风气候,加工工艺的不同和气候的不同很可能导致了香肠细菌分布的不同,进而导致了香肠风味的不同。
在细菌水平上,4个地区中厚壁菌门,蓝藻门和变形菌门占绝对优势,其中SC地区乳杆菌属和葡萄球菌属占优势,GD地区中乳球菌属,不动杆菌属和巨型球菌属含量较高,HN地区中乳球菌属,四联叠球菌属,乳杆菌属和巨型球菌数含量较高,前三者都是属于乳酸菌,HB地区中,D1样品中的类芽孢杆菌属含量较高,剩下的样品相对于无法比对的细菌来说含量并不是太高,但从中可以看出乳酸菌含量还是具有一定比例。乳酸菌在发酵肉制品中占优势,和前人的研究结果是一致的[19-21],其在发酵肉制品中主要是通过酸化来改变产品的品质[22]。巨型球菌属和葡萄球菌属在基因上相比[23],具有相近的进化关系,对4个地区检测到的巨型球菌属进一步在菌种水平划分,确定该菌为溶酪大球菌。溶酪大球菌呈革兰氏阳性,早期研究表明该菌种作为内源发酵剂,可提高对脂肪和蛋白质的分解作用,并可以耐受一定浓度的食盐,具有亚硝酸盐分解能力,并对改善香肠的风味具有一定的积极作用[24-26],对生物体没有致病作用[27],通过全基因测序发现,该菌种和葡萄球菌在代谢途径上极为相似,但是和葡萄球菌相比,该菌种的染色体较小并且缺少毒力基因[23]。但近期研究表明,该菌的某些菌株对禽类具有感染性[28],为了食品安全,还需进一步对该菌种做进一步的研究。
在传统发酵产品中,本研究结果中微生物多样性特征和其他学者相比较,大体上是一致的但也有部分差异,例如在传统发酵肉制品中,乳酸菌和凝固酶阴性球菌是两类主要的菌群[29],本研究结果中除了这两类细菌之外,蓝藻门也占据了一定比例。引起这种差异可能有以下几个原因:采样的时间、气候,制作香肠的工艺,制作香肠原料肉的肥瘦比,添加香辛料等的不同造成微生物生存环境的不同,从而造成了这种差异。本研究中存在的未测定微生物可能是因为宏基因组测序数据中含有多种未知物种,从而很难确定一段DNA序列来源于何种物种[30]。本课题组选取了4个地区共20种香肠进行研究,虽然有一定的局限性,但也能够反映中式香肠细菌群落分布。
