超高效液相色谱-四极杆/静电场轨道阱高分辨质谱快速分析化妆品中17种喹诺酮类药物
2019-09-27廖华勇李红玉
廖华勇,王 景,李红玉
(广州华鑫检测技术有限公司,广东 广州 510663)
喹诺酮类药物(Quinolones,QNs)是一类具有4-喹诺酮结构的合成抗菌类药物,因具有抗菌谱广、杀菌能力强、价格低、与其他抗菌药物无交叉耐药性等特点,被广泛用于由细菌和支原体引起的人和动物的疾病治疗[1-2]。化妆品作为人们日常生活的必需品,一直受到全世界人民的关注。特别是随着生活水平的提高以及消费能力的增强,越来越多的人群开始注重皮肤保养,这就对化妆品的质量及功效提出了更高要求。然而,部分厂家为增强产品祛痘、除螨和抗粉刺的效果,在化妆品中添加喹诺酮类药物。该类药物可通过皮肤微血管和黏膜吸收,长期不当或过量使用可能导致其在人体蓄积,进而破坏皮肤表层菌群,导致皮疹、过敏和病原体产生抗药性等损害。我国《化妆品安全技术规范》(2015年版)[3]及欧盟化妆品法规(EC)No.1223/2009[4]均明确规定不得将抗生素类药物作为化妆品的生产原料及添加组分。因此,建立一种灵敏、快速、准确的化妆品中喹诺酮类药物的检测方法对于保障化妆品质量安全具有重要意义。
目前,喹诺酮类药物的检测方法主要有酶联免疫法(ELISA)[5]、胶体金免疫层析法(GICA)[6]、电化学法(EC)[7]、毛细管电泳-电化学发光法(CE-ECL)[8]、高效液相色谱法(HPLC)[9-11]、液相色谱-串联质谱法(LC-MS/MS)[12-15]、液相色谱-高分辨质谱法(LC-HRMS)[16-18]等。由于化妆品种类多、基质差异大,且违法添加的抗生素药物含量低,这不仅要求样品提取与净化技术具有普适性和高效性,还要求测试方法具有良好的选择性和高灵敏度。上述已报道的ELISA、EC、HPLC、LC-MS/MS等方法虽然可对喹诺酮类化合物进行检测,但在选择性和阳性确证方面仍存在不足。HRMS具有高通量、高选择性和高灵敏度等特点,抗干扰能力强,可适用于基质复杂、待测物含量低的样品分析。近年来,液相色谱-高分辨质谱联用法越来越多地应用于化妆品等复杂基质中药物和非法添加物质的多组分分析[16-18]。本文采用简单的正己烷液液萃取净化结合超高效液相色谱-四极杆/静电场轨道阱高分辨质谱(UPLC-Q Orbitrap HRMS),建立了一种同时测定水剂、乳液和非蜡基膏霜类化妆品中17种喹诺酮类药物的检测方法。该方法操作简单,定性定量分析准确,适用于化妆品中喹诺酮类药物的安全性评价分析。
1 实验部分
1.1 仪器与试剂
Ultimate 3000 超高效液相色谱仪和Q-Exactive四极杆/静电场轨道阱高分辨质谱仪(美国Thermo公司);TurboVap LV氮吹仪(美国Caliper公司);KQ-500E数控超声波清洗器(昆山超声仪器有限公司);KDC-1044离心机(中科佳仪有限公司);Milli-Q超纯水器(美国Millipore公司);MS3涡旋仪(德国IKA公司)。
标准品:氧氟沙星、恩诺沙星、环丙沙星、诺氟沙星、二氟沙星、培氟沙星、沙拉沙星、司帕沙星、丹诺沙星、氟罗沙星、奥比沙星、麻保沙星、依诺沙星、洛美沙星、加替沙星、西诺沙星和那氟沙星,纯度均大于98%,购自德国Dr.Ehrenstorfer公司;内标:D5-恩诺沙星(纯度大于99%,德国Witega公司);乙腈、甲醇、正己烷(色谱纯,德国Merck公司);甲酸(色谱纯,美国Sigma公司);超纯水(18.2 MΩ·cm,由Millipore纯水仪制备)。
样品为随机的市售水剂、乳液和非蜡基膏霜类化妆品。
1.2 标准溶液配制
100 mg/L单标储备液:分别准确称取各标准品10 mg于100 mL棕色容量瓶中,以50%乙腈溶液(含1%甲酸)定容至刻度,-18 ℃储存。
100 μg/L混合标准工作溶液:准确移取各单标储备液0.25 mL于250 mL棕色容量瓶中,以50%乙腈溶液定容至刻度,使用前现配。
1.0 mg/mL内标储备溶液:准确称取D5-恩诺沙星10 mg于10 mL棕色容量瓶中,以50%乙腈溶液(含1%甲酸)定容至刻度,-18 ℃储存。
1.0 mg/L内标工作溶液:准确移取内标储备溶液0.1 mL于100 mL棕色容量瓶中,以50%乙腈溶液定容至刻度,-18 ℃储存。
1.3 样品前处理
称取试样1.0 g(精确至0.001 g),置于25 mL具塞比色管中,加入1.0 mg/L内标工作溶液0.1 mL,涡旋混匀,加入20 mL 0.1%甲酸乙腈溶液,涡旋振荡1 min,超声提取20 min,以0.1%甲酸乙腈溶液定容至25 mL,摇匀。移取2.0 mL上述提取液,经10 000 r/min高速离心2 min后,准确移取1.0 mL上清液于50 ℃水浴中氮吹至近干,用10%乙腈溶液定容至2.0 mL,加入2 mL乙腈饱和的正己烷溶液,涡旋振荡1 min后,4 000 r/min离心2 min,下层清液经聚四氟乙烯滤膜过滤后待测定。
1.4 检测条件
液相色谱条件:Agilent Poroshell EC C18(4.6 mm×150 mm,2.7 μm)色谱柱;柱温:30 ℃;流动相:A为0.1%甲酸乙腈,B为0.1%甲酸水,梯度洗脱条件:0.0~8.0 min,10%~30%A;8.0~8.1 min,30%~10%A;8.1~12.0 min,10%A;流速:0.3 mL/min;进样量:5.0 μL。
质谱条件:加热电喷雾离子源(HESI)温度为350 ℃;毛细管电压为3.5 kV;离子传输管温度为320 ℃;鞘气压力为 0.276 MPa,辅助气流速为 3.3 L/min;全扫描/二级离子扫描(Full Scan/dd-MS2)模式:采集范围为m/z100~600,正离子采集;一级质谱分辨率为70 000 FWHM,二级质谱分辨率为17 500 FWHM;归一化碰撞能量(NCE)为17.5、35.0、52.5 eV。其他分析参数见表1。

表1 17种喹诺酮类药物的分析参数Table 1 Analysis parameters of 17 quinolones
2 结果与讨论
2.1 检测条件的优化
2.1.1 色谱柱的选择考察了Agilent Poroshell EC C18(4.6 mm×150 mm,2.7μm)、Waters ACQUITY UPLC BEH(2.1 mm×100 mm,1.7 μm)和Thermo Hypersil GOLD C18(2.1 mm×100 mm,1.9 μm)3种液相色谱柱对17种喹诺酮类化合物的分离效果。结果显示:后2种色谱柱对目标化合物的保留相对较差,色谱峰的半峰宽较宽,丹诺沙星、司帕沙星等部分化合物的峰形出现拖尾;而所有目标化合物在Agilent Poroshell EC C18色谱柱上均能实现有效保留,且重现性好,因此选择该色谱柱对样品进行分离。
2.1.2 流动相的选择喹诺酮类化合物的母核结构为氧代喹啉羧酸,含有羧基和氮原子,因此在酸性条件下有利于形成[M+H]+峰。为增加化合物响应,实验所用流动相中均加入0.1%甲酸。首先选取0.1%甲酸乙腈为有机相,考察了0.1%甲酸水、5 mmol/L 乙酸铵(含0.1%甲酸)为水相时的分离效果。结果表明:0.1%甲酸水和5 mmol/L乙酸铵(含0.1%甲酸)两种水相下化合物的出峰响应相差不大(5%以内),但0.1%甲酸水条件下化合物的峰形对称,因此选择水相为0.1%甲酸水溶液,进一步比较了0.1%甲酸乙腈和0.1%甲酸甲醇分别为有机相时的分离效果。结果显示,两种有机相下目标物的质谱响应和峰形相当,但有机相为乙腈时,色谱柱柱压低。为有效保护色谱柱,实验最终选择0.1%甲酸乙腈与0.1%甲酸水为流动相。在优化条件下,17种喹诺酮类化合物的响应和峰形均满足分析要求(见图1)。


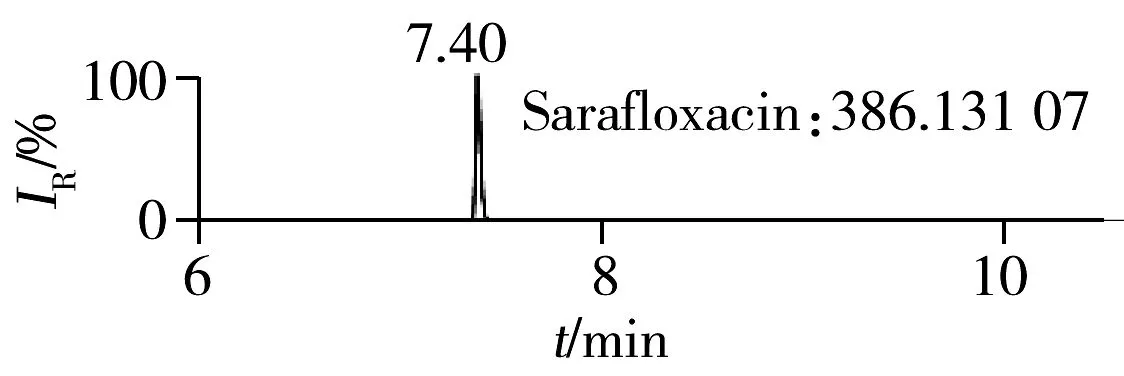
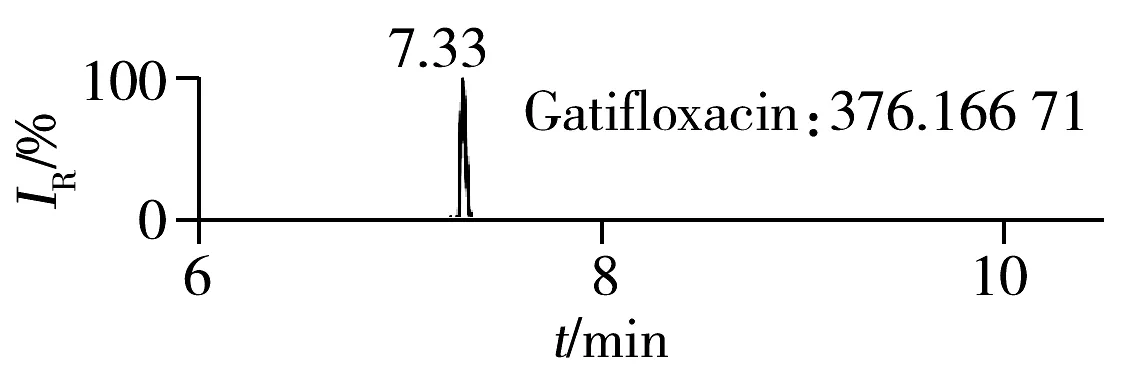

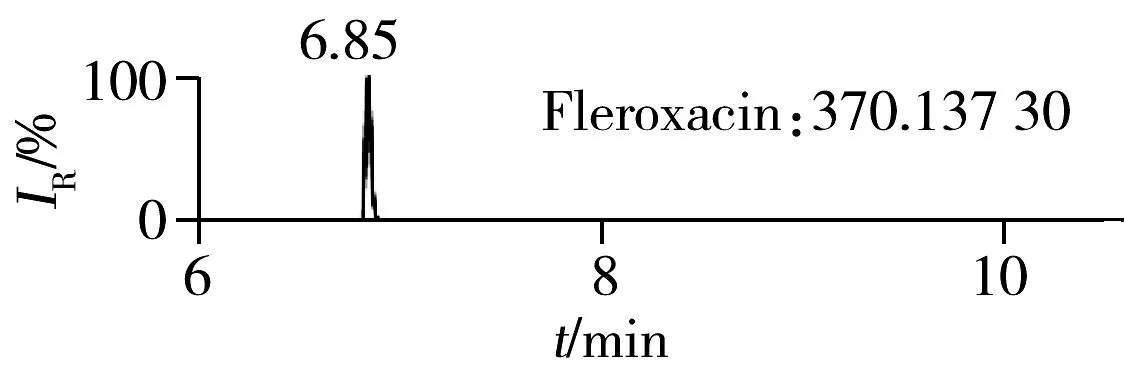
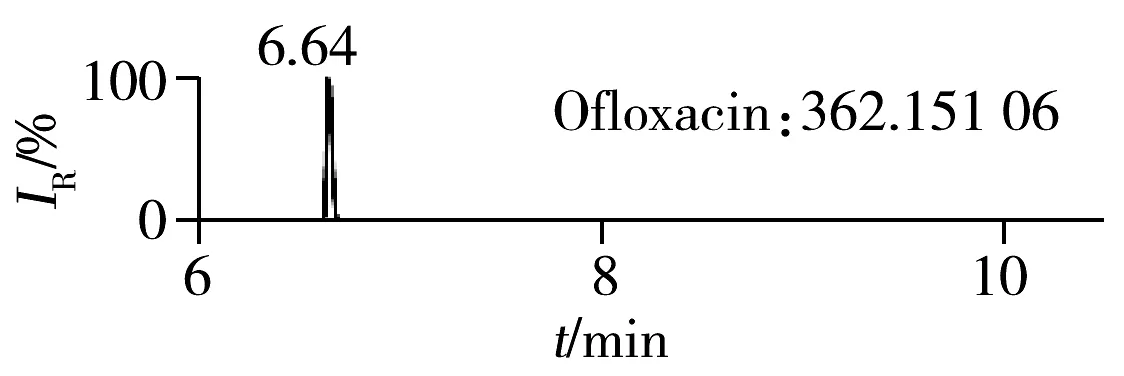

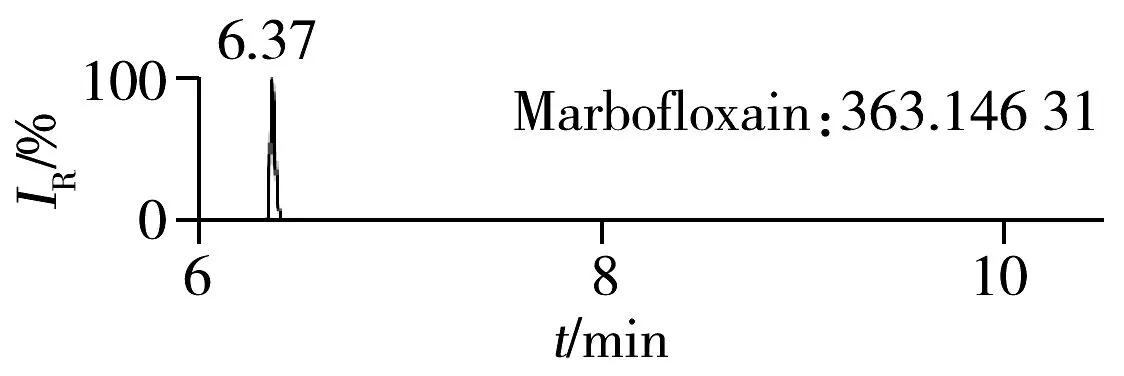

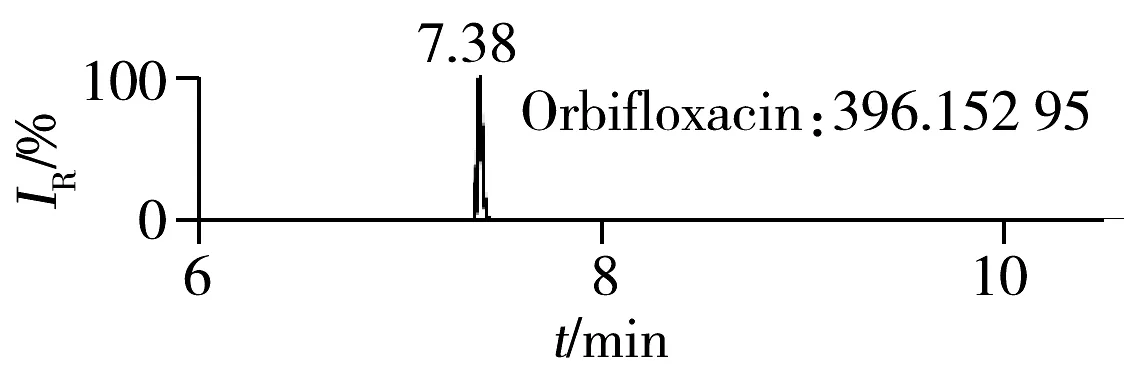
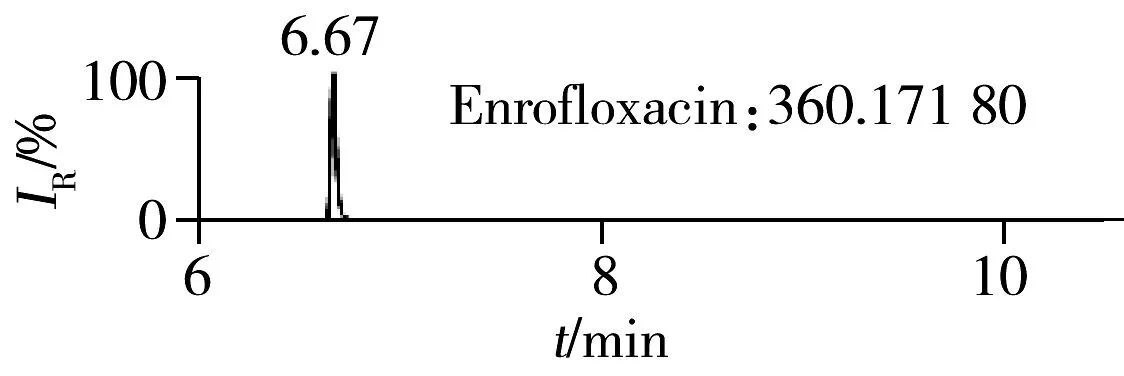
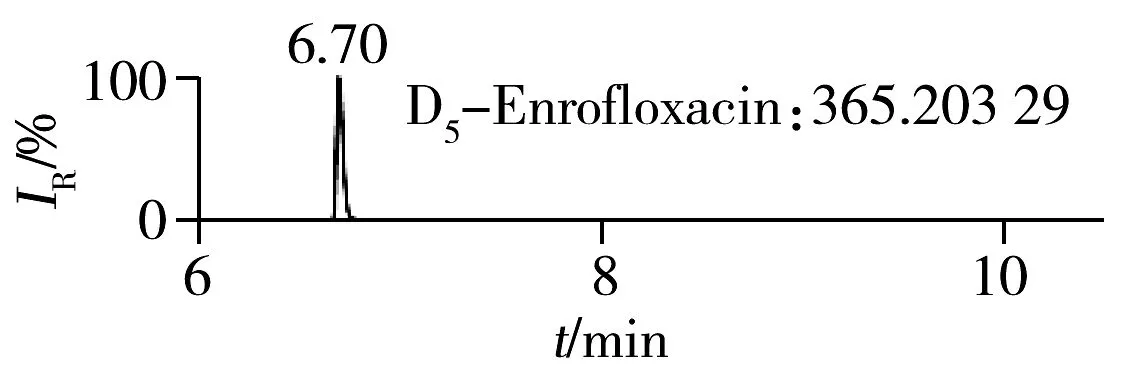
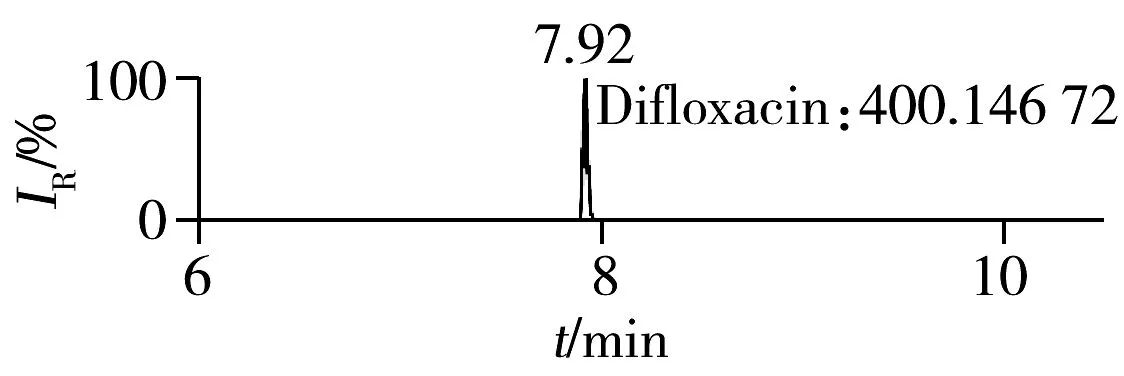
图1 喹诺酮类化合物的提取离子流色谱图Fig.1 Extracted ion chromatograms of quinolones
2.2 样品前处理方法的优化
2.2.1 提取方式的优化常用的化妆品样品提取方式包括超声萃取、涡旋振荡和水平振荡。由于膏霜类化妆品的黏度较大,在涡旋振荡和水平振荡过程中常出现分散不均匀现象。而超声萃取法利用超声波的空化作用、机械效应和热效应来加速样品中各组分的释放,使其有效扩散和溶解于提取溶剂中,从而显著改善提取效果,适合于膏霜类化妆品的前处理。实验考察了不同超声时间(5、10、15、20、30 min)对加标100 μg/kg的非蜡基膏霜类化妆品中目标物提取效率的影响,结果表明,随着超声时间的延长,目标物的提取率逐渐增加,超声时间为20 min时提取率达到最大,继续延长超声时间,提取率无显著提高。因此,选择超声提取20 min。
2.2.2 提取溶剂的选择由于喹诺酮类化合物易溶于有机溶剂,可采用乙腈进行萃取。考虑到不同化妆品的基质特点,分别以不同体积分数的乙腈水溶液(100%、80%、50%、20%)作为提取溶剂,对加标100 μg/kg的非蜡基膏霜类化妆品进行提取。结果显示,提取溶剂中有机相比例≥80%时,17种化合物的提取回收率较高,可达80%以上。为便于后续氮吹浓缩,实验选取100%乙腈为提取溶剂。此外,根据喹诺酮类化合物的pKa可知,其在酸性溶剂中溶解度更好,因此最终以0.1%甲酸乙腈为提取溶剂。
2.2.3 净化方法的选择喹诺酮类药物的非法添加量一般较小,若提取液直接使用高分辨质谱测定,则化妆品中含有的大量脂类和表面活性剂等物质会对目标化合物的检测产生严重干扰,因此需先对提取液进行净化。目前,化妆品的常用净化方法有正己烷液液萃取脱脂、分散固相萃取净化及固相萃取柱净化等。实验考察了HLB固相萃取柱、PSA+C18分散固相萃取以及正己烷液液萃取脱脂3种净化方式对加标100 μg/kg的非蜡基膏霜类化妆品的提取效果。结果表明,选取HLB固相萃取柱净化时,目标化合物的回收率为80%~110%,但净化步骤繁琐,操作复杂;采用PSA+C18分散固相萃取和正己烷液液萃取脱脂净化时,目标化合物的回收率同样能达到80%~110%,为节约成本和便于操作,最终选择正己烷液液萃取脱脂净化。
2.3 方法验证
2.3.1 线性关系、检出限与定量下限本实验以D5-恩诺沙星为内标,采用内标法定量,以校正ESI离子源在检测样品时出现的基质效应。在优化条件下,采用本方法分别测定质量浓度为0.05~20.0 μg/L的系列混合标准工作溶液,其中D5-恩诺沙星的质量浓度为4.0 μg/L。以各化合物与内标物的质量浓度比值为横坐标(x),以化合物与内标物的响应峰面积比值为纵坐标(y)进行线性回归。结果表明,17种喹诺酮类化合物均在0.05~20.0 μg/L范围内线性关系良好,相关系数(r2)均大于0.996 0(见表2)。
通过测定质量浓度为1.0 μg/L的混合标准溶液响应值,分别以3倍信噪比(S/N=3)和10倍信噪比(S/N=10)计算得到各化合物的仪器检出限(ILOD)和仪器定量下限(ILOQ)。结合前处理过程中的稀释倍数(n)和回收率(R),计算得到17种喹诺酮类化合物的方法检出限(LOD=ILOD×n/R)和定量下限(LOQ=ILOQ×n/R)分别为0.5 μg/kg和1.0 μg/kg。
2.3.2 回收率与相对标准偏差在优化条件下,选取2种基质(爽肤水、面霜)的阴性样品进行加标回收实验。每种基质分别添加2、4、20 μg/kg 3个浓度,每个浓度平行测定6次,计算回收率和相对标准偏差(RSD)。结果表明,3种加标水平下的回收率为82.3%~108%,RSD为3.5%~8.6%(见表2)。方法的准确度和精密度均能满足日常检测定量分析的要求。
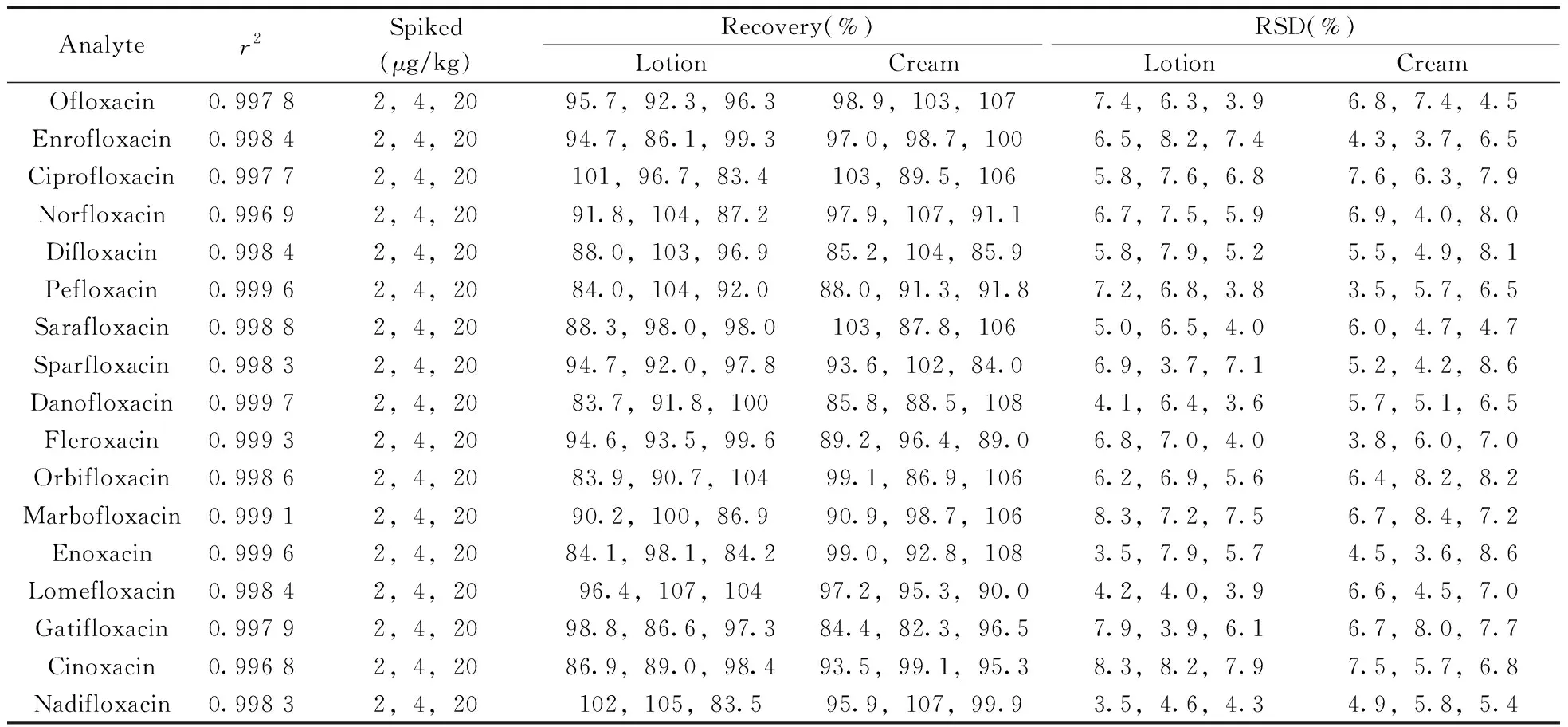
表2 17种喹诺酮类化合物的相关系数(r2)、加标回收率及相对标准偏差(n=6)Table 2 Correlation coefficients(r2),spiked recoveries and RSDs of 17 quinolones(n=6)

图2 阳性样品中氧氟沙星的提取离子色谱图Fig.2 Extracted chromatogram of ofloxacin in a positive sample
2.4 实际样品的测定
为评价该方法的有效性,本实验测定了随机抽取的市售水剂、乳液和非蜡基膏霜类化妆品各10份,在1份标称具有除痘功效的膏霜样品中检出氧氟沙星,检出量为1.7 mg/kg;其他29份样品均未检出目标化合物。阳性样品的提取离子色谱图见图2。
3 结 论
本文利用0.1%甲酸乙腈进行超声提取,正己烷液液萃取脱脂净化,建立了化妆品中17种喹诺酮类化合物的超高效液相色谱-四极杆/静电场轨道阱高分辨质谱检测方法。通过对色谱柱、流动相等分离条件和样品提取净化条件的优化,得到了良好的加标回收率和精密度,方法检出限和定量下限分别为0.5 μg/kg和1.0 μg/kg。该方法前处理简单、适用性好,可为化妆品质量监管和控制提供技术支持。
