共检猪流行性腹泻病毒、猪传染性胃肠炎病毒和猪轮状病毒的cDNA 芯片的技术优化
2019-09-17胡靖飞尹人杰黄小波刘志鹏赵玉佳曹三杰文心田文翼平
胡靖飞,尹人杰,黄小波,2,3*,刘志鹏,赵玉佳,曹三杰,2,3,文心田,文翼平,赵 勤,伍 锐
(1.四川农业大学动物医学院猪病研究中心,成都 611130;2.农业部兽用药物与兽医诊断技术四川科学观测实验站,成都 611130;3.四川农业大学国家级动物类实验教学示范中心,成都 611130)
猪流行性腹泻病毒(porcine epidemic diarrhea virus,PEDV)、 猪传染性胃肠炎病毒(transmissible gastroenteritis virus,TGEV)和 A 型猪轮状病毒(porcine rotavirus-A,PoRVA)是三种主要引起哺乳仔猪发生腹泻、呕吐、脱水等症状的病毒,中大猪多呈隐形感染,2 周龄以内的小猪发病率高, 致死率较高,其中PEDV 引起的死亡率高达70%~100%[1-3]。三者的流行病学、发病症状和病理病变相似,难以区分,特别是生产中三种病毒常呈混合感染。目前实验室诊断包括分子生物学诊断及免疫学诊断, 主要有RT-PCR[4]、LAMP[5]、ELISA[6]、免疫荧光和病毒分离鉴定[7]等。但是这些方法在临床应用中均存在耗时费力,程序复杂,准确性不高易出现假阳性,无法同时高通量鉴定混合感染等问题,因此,需要开发一种能同时对混合感染进行高通量、快速便捷的检测方法。
基因芯片技术的原理是通过碱基互补配对原则将固定在载体上的探针与样品cDNA 进行杂交,以标记的信号报告分子获得杂交信号[8]。T.R.Gingeras 等[9]建立了高密度DNA 芯片同时鉴定分枝杆菌菌种和结核分枝杆菌耐利福平rpo B 基因突变。T.L.Nicholson 等[10]针对猪繁殖与呼吸综合征病毒、猪圆环病毒2 型和猪呼吸道冠状病毒设计了检测基因芯片。N.Y.Park 等[11]建立了cy5 间接标记的能同时对 PEDV、TGEV、PECV、PoRV A 群和 C 群等造成猪腹泻病病毒的检测芯片。
本实验室前期滑翔等[12]用间接荧光标记技术和多重RT-PCR 技术相结合,建立了检测3 种猪病毒性腹泻的cDNA 基因芯片,马锐等在此基础上又对靶基因的直接荧光和间接荧光标记方式进行比较研究, 结果显示引物直接荧光标记法杂交的信号值、信噪比和灵敏度都优于荧光间接标记法[13]。但仍存在部分缺陷,如点制过程不稳定造成探针点大小不均、洗涤处理不彻底、常规PCR 扩增标记产生单链有限,杂交信号不稳定等缺陷。为提升该病毒腹泻cDNA 芯片的检测效果与稳定性,建立标准的芯片制备方法,本研究在前期滑翔等构建的cDNA 芯片基础上, 重点样品标记技术方面进行了改进,并对芯片制作与杂交的关键条件进行了优化研究,确定了PEDV-TGEV-GAR 共检cDNA 基因芯片技术的最佳参数,为后续基因芯片的标准化制备与应用奠定基础。
1 材料与方法
1.1 仪器及试剂
基因芯片点样仪及扫描仪购自北京博奥生物公司;PCR 仪和分子杂交仪购于美国Thermo 公司;氨基载玻片、 点样缓冲液购自上海百傲生物公司;质粒提取试剂盒购于美国Omega 公司;RNA 提取试剂盒购自上海生工公司;反转录试剂盒和λ 定位基因购自 Takara 公司;2×Taq PCR Master Mix 购自天根生物公司;pMD-PEDV-S、pMD-PEDV-M、pMD- TGEV-S、pMD-TGEV-N、pMD-GAR-VP7、pMD-GAR-NSP4 重组质粒菌, 均由四川农业大学猪病研究中心构建保存。
1.2 引物设计
定位基因引物参考曹三杰等[14]设计合成。探针及靶基因引物参照滑翔[12]等设计的6 对引物,退火温度在(55±2)℃范围内,由上海生工生物公司合成,其中反向引物为两条,扩增靶基因引物合成是在5′端用Cy3 修饰,扩增探针的引物不作修饰(表1)。
1.3 基因芯片的设计和制备
将含探针基因的重组质粒菌 (pMD-PEDV-S、pMD -PEDV -M、pMD -TGEV -S、pMD -TGEV -N、pMD-GAR-VP7、pMD-GAR-NSP4)复苏培养,抽取质粒作为 PCR 扩增模板。PCR 体系(50 μL)为:2×Taq PCR Master Mix 25 μL、 上下游引物各 2.0 μL、质粒及定位基因 2.0 μL、ddH2O 补足至 50.0 μL。PCR程序为:94 ℃ 2 min;94 ℃ 30 s,54 ℃ 30 s,72 ℃ 30 s,34 个循环;72 ℃ 10 min;4 ℃保存。产物用乙醇沉淀法纯化后作为探针, 并与点样缓冲液按1∶1 混合加入386 孔板,阴性对照为点样液,按设计好的矩阵参数进行接触式点样(图1)。点样结束后再经过烘干、水合、紫外交联和水洗4 个步骤,完成芯片的构建。并将构建好的芯片真空抽干,于4 ℃保存。

表1 靶基因及探针扩增引物Table 1 Primers for target genes and probes amplification

图1 基因芯片矩阵设计图Figure 1 The design of DNA microarray spotting
1.4 芯片制备最佳条件优化
1.4.1 点样缓冲液的选择及浓度优化
由于点样液为购买商品化, 成分公司未公开,因此不得而知,在使用过程中发现单独使用效果不够理想,因此尝试混合使用,选取博奥点样液(CB)、百傲点样液(BO),两者分别按 1∶1、1∶2、2∶1 混合(点样液中混有荧光),再分别与PM 探针按1∶1 比例混合。用点样仪点样,置芯片扫描仪扫描。
1.4.2 水合时间优化
将点制好的芯片置于80 ℃的杂交仪中(或密闭盒子放于烘箱中),烘干 8 h 后,分别按 0、2、4、6 s水合处理。紫外交联30 min 后,置芯片扫描仪扫描分析。
1.4.3 探针基因浓度优化
用点样缓冲液将探针 PM(1.29×1010拷贝/μL)分别稀释至 800、600、400、200、100、50 ng/μL,将不同浓度的探针基因分别点制在芯片上。再与PM 靶基因杂交,扫描分析结果,确定探针最佳点样浓度。
1.4.4 洗液及干燥方式优化
将芯片分别置于0.2%SDS 和超纯水中上下抽提5 min,再将清洗后的芯片分别用离心机1 000 r/min甩干和自然晾干。
1.5 不对称PCR靶基因引物比例优化
以 PM、PS、TN、TS、NSP4,VP7 质粒为模板,以5′端标记有Cy3 的反向引物和非荧光正向引物,按照浓度比例为 1∶10、1∶20、1∶40、1∶50 进行不对称PCR 扩增,同时设立 1∶1 比例的常规 PCR 扩增作对照组。反应体系及程序同1.3,扩增产物避光保存备用。取上述扩增产物,高温变性后,与杂交缓冲液按1∶1 体积混合, 取混合液 50 μL 加入芯片区, 按照1.3 操作步骤杂交完毕后,立即洗涤,甩干、扫描分析。
1.6 杂交条件优化
将1.5 中扩增好的靶基因与杂交缓冲液以1∶1的比例混合, 然后将混合液于95 ℃变性3 min,迅速置于冰上冷却后取50 μL 加到阵列上,置于杂交仪内。(此步骤后避光操作)。选取同一批次制备的相同芯片,分别进行杂交时间和杂交温度的优化。
1.6.1 杂交时间优化
在48 ℃下与芯片杂交, 杂交时间分别为1、2、3、4、5 和6 h,扫描分析后确定最佳杂交时间。
1.6.2 杂交温度优化
使杂交盒中靶基因产物在 40、44、48 和 52 ℃温度下杂交3 h,扫描分析确定最佳杂交温度。
1.7 优化后与前期芯片效果比较
将条件优化后的芯片与前期制备的芯片按各自操作程序检测已知PEDV 病毒核酸阳性的病料,比较杂交后效果。
1.8 共检芯片的检测效果验证
根据上述优化后的最佳参数点制芯片,以5 份已知感染背景的临床病料样品,按芯片检测流程进行检测,验证该芯片的检测效果。
2 结果
2.1 探针制备结果
提取靶基因重组质粒, 扩增获得了与前期滑翔等报道大小一致的基因片段:PEDV-M:421 bp、PEDV-S:474 bp、TGEV-N:362 bp、TGEV-S:276 bp、GAR-NSP4:270 bp、GAR-VP7:381 bp(常规 PCR 图此处略)。扩增产物纯化后即可用作芯片点样探针。
2.2 芯片制备最佳条件优化结果
2.2.1 点样缓冲液的选择及浓度优化
选取的两种不同点样液按不同比例混合后点样,扫描分析。结果显示,单独使用CB 时,样点小扫描效果不佳;单独使用BO 时,样点边缘不规则且扩散不均。两者按2∶1 混合点样,样点扩散不均匀,两者按1∶1 混合,样点大小合适,且扩散均匀。因此在点样时选取CB 与BO 按 1∶1 混合后加入探针(图 2)。
2.2.2 水合时间优化
比较不同水合时间,水合时间在0 s 时,样点的大小不均匀;而超过4 s 以后,样点扩散过快,造成各样点之间出现的粘连情况,不利于芯片杂交及扫描。水合4 s 比2 s,样点更均匀,且扩散效果更好(图 3)。

图2 点样缓冲液优化结果Figure 2 Optimization results of printing buffer
2.2.3 探针基因浓度优化
探针基因浓度优化结果表明(图4),当探针基因的浓度在 100 ng/μL 和 50 ng/μL 时 SNR<2,无法判为阳性;在200 ng/μL 时,肉眼观察斑点并不能准确判定为阳性;而在 600~800 ng/μL 中,阳性标记靶基因均能在杂交信号和肉眼观察下直观反映。综合芯片制作成本, 本研究确定600 ng/μL 作为所有探针基因的固定浓度。
2.2.4 洗液及干燥方式优化
用0.2% SDS 进行清洗时, 对未固定上的探针清洗更为彻底,但相对来说背景值稍有提高。而在用超纯水清洗时,背景值变化不大,但清洗效果不如用0.2% SDS。而在用自然晾干的方式进行干燥处理时,背景值显著提高,因此选用0.2% SDS 清洗,且 1 000 r/min 离心甩干(图5)。
2.3 不对称PCR靶基因引物比例优化
选择 1∶1、1∶10、1∶20、1∶40、1∶50、1∶60、 1∶80、1∶160、1∶200 比例进行不对称PCR 扩增,同时与常规PCR 扩增作对照,杂交扫描分析(图6)。由表2 可知,比例在 1∶20 与 1∶40 杂交效果均优于常规PCR。综合结果与成本, 选取1∶40 比例的不对称PCR 扩增来标记靶基因。
2.4 杂交条件优化
2.4.1 杂交时间优化

图3 水合时间的选择结果Figure 3 Selection of hydration time

图4 不同浓度探针结果Figure 4 The image of different concentration probes
杂交结果显示,杂交时间在1~6 h 之间,均可判定为阳性杂交成立。且每个探针基因的信号中位值随杂交时间而增加,而背景值也相应增加。综合考虑,杂交时间选择3 h(图7)。
2.4.2 杂交温度优化

图5 洗液及干燥方式的选择结果Figure 5 Selection of washing liquor and drying
图6 不同比例不对称PCR 扩增杂交Figure 6 Different concentration ratio of the primers to asymmetric PCR
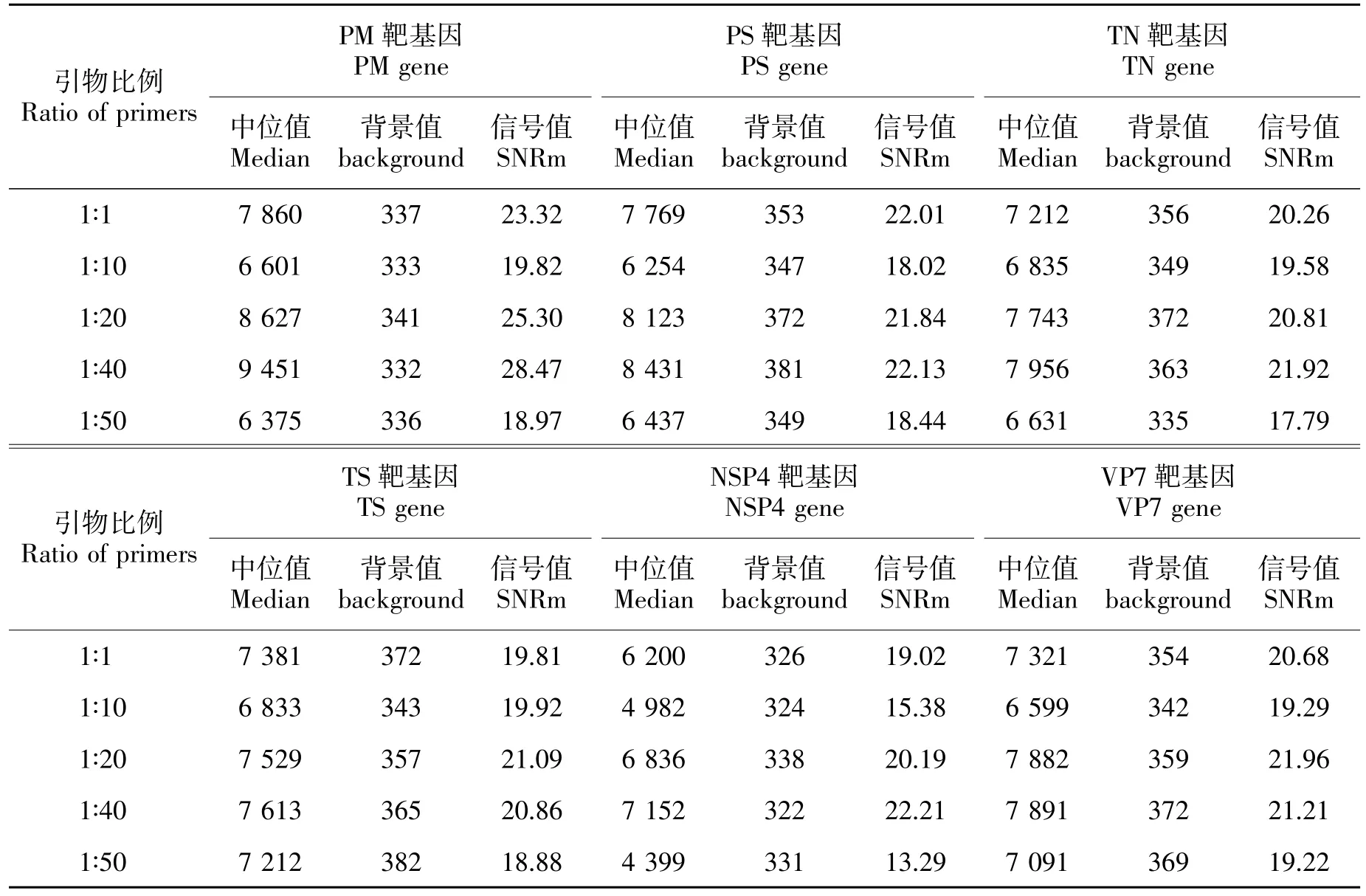
表2 不同比例不对称PCR 荧光信号扫描结果Table 2 Signal intensity of different concentrations of asymmetric PCR
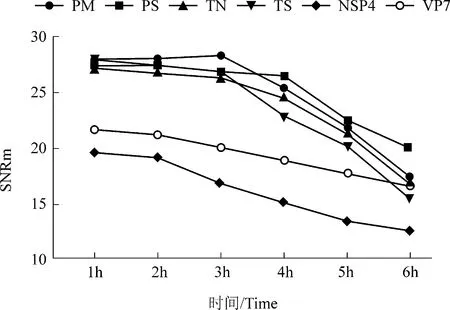
图7 不同杂交时间的芯片结果Figure 7 Results at different hybridization time
由表3 及图8 可知,随着杂交温度升高,信号随之增强,同时背景值也会增强,在52 ℃时的背景值过大。在 40 ℃和 44 ℃时 NSP4 和 VP7 探针的信号值与其他探针差异较大, 而48 ℃时各探针信号差异较小, 因此选择48 ℃为共检芯片的最佳杂交温度。
2.5 优化前后芯片检测效果对比
如图9 所示,优化前的芯片,样点大小不均匀,杂交效果不理想,荧光重复性较差;相比而言,优化过后样点大小均一,荧光重复性好。

图8 不同温度的杂交结果Figure 8 Hybridization result at different temperature
2.6 共检芯片检测效果的验证
以确定的最佳参数条件构建芯片,选取的5 份临床样品进行效果验证,以反转录的cDNA 为模板进行不对称PCR 扩增标记,再与芯片杂交,结果显示检测临床样品效果可靠, 与PCR 检测结果一致(图10)。

表3 不同杂交温度的荧光信号扫描结果Table 3 Signal intensity at different hybridization temperatures

图9 优化前后效果比较Figure 9 DNA chip compared before and after optimization.
3 讨论
本团队前期滑翔等[12]构建了共检PEDV、TGEV和PRoV 的cDNA 基因芯片, 本研究在重点在样品标记技术方面进行了创新与改进,采用直接荧光标记与不对称PCR 技术相结合提高芯片杂交信号,同时还对芯片制作与杂交的多个关键技术环节进行了优化研究。目前较常用的芯片载体包括膜片和玻片两种, 玻片依赖于表面修饰的化学基团与DNA形成Schiff 碱达到固定探针的作用[15-16],本研究采用氨基硅化处理的玻片作为芯片的载体基片。烘焙和紫外交联是为了让探针基因更好的与基片表面共价结合。前期样点大小不均或出现粘连情况,会使得扫描出的数据会有很大的误差,本研究对点样缓冲液的比例及水合时间进行了逐一优化,很好的控制了阵点的大小分布。探针基因浓度主要影响信号强度,靶基因量过剩,探针浓度越高信号强度则越趋于饱和,反之信号强度则越弱。

图10 样本检测结果Figure 10 The detection results of known samples.
N.Y.Park 等[11]建立的对猪腹泻病毒共检的寡核苷酸芯片采用CY5 标记dCTP 进行PCR 扩增,但CY5 对空气中的O3比较敏感, 暴露在空气中的CY5 易被其破坏,对结果分析会造成一定干扰[17],因此本研究选择CY3 进行标记[18]。本实验室前期对靶基因的直接标记法和间接标记法进行了对比,结果表明直接标记法的灵敏性是间接标记法的100 倍[13],所以直接采用了直接标记法对靶基因下游引物标记。前期采用多重PCR 方法进行靶基因的扩增,获得的靶基因单链有限。Kawai 的研究表明芯片杂交时,靶基因双链中起杂交作用的实际上只是其中的一条链,而另一条链对杂交有一定抑制作用,使靶基因以双链的杂交效率低于靶基因单链的杂交效率[19]。本研究以不对称PCR 扩增靶基因,目的是得到单链的杂交模板,进一步提高杂交效率。不同靶基因扩增的引物的不对称比例有所不同,这与扩增产物片段大小和引物引导的扩增的效率有一定关系[20]。通过比较上下游引物比例 1∶10~1∶200,从芯片杂交分析得出了正反向引物的最适比例为1∶40。并非单链量多则信号值高,双链量多则信号值低。这与不对称PCR 扩增出单链和双链比例有很大的关系。因此在以提高芯片杂交效率前提下,单独判断不同引物比例产生单链和双链量无意义,而要综合其扩增靶基因产物与芯片杂交效率之间的关系。杂交温度和杂交时间是影响杂交效果的两个重要参数,本研究进行了两个参数的筛选,综合比较各杂交条件的结果,发现升高杂交温度或是延长杂交时间都会使杂交信号呈现上升趋势, 但是温度过高、时间过长造成背景值升高,这是可能是由于杂交液随时间延长蒸发过多,从而造成了背景值的升高,影响扫描仪对信号获取[21]。同时,由于每种探针的序列不一致,最适杂交温度也会有差异。基于这两点考虑通过筛选优化,使中位值与背景值的比值即SNRm 值达到最大。
通过对3 种猪病毒性腹泻共检芯片的各项参数进行优化选择,同时以荧光直接标记引物结合不对称PCR 等方法, 进一步提高该共检芯片的检测效果,为开展芯片的标准化制作和临床应用奠定了基础。
