Amino functionalized chiral mesoporous silica nanoparticles for improved loading and release of poorly water-soluble drug
2019-09-12XinWngChngLiNFnJingLiHotinZhngLeiShngZhongguiHeJinSun
XinWng,ChngLi,NFn,JingLi,HotinZhng,LeiShng*,ZhongguiHe,JinSun,d,**
a Wuya college of Innovation, Shenyang Pharmaceutical University, No.103, Wenhua Road, Shenyang 110016, China b College of Basic Medical Science, Shenyang Medical College, No. 146 Huanghe North Street, Shenyang 110034,China c School of Life Science and Biopharmaceutics, Shenyang Pharmaceutical University, No.103, Wenhua Road,Shenyang 110016, China d Municipal Key Laboratory of Biopharmaceutics School of Pharmacy, Shenyang Pharmaceutical University, No. 103,Wenhua Road, Shenyang 110016, China
Keywords:Chiral mesoporous silica Amino functionalization Curled drug loading Curled drug release
ABSTRACT In the present paper, chiral mesoporous silica nano-cocoon (A-CMSN) functionalized with amino group was synthesized, and its loading and release of indomethacin (IMC), a poorly soluble drug, was studied. Due to the use of chiral anionic surfactants as a template, ACMSN possessed 2D hexagonal nano-cocoon morphology with curled channels on its surface, which was quite different from another 2D hexagonal mesoporous silica nanoparticles(MCM-41) with straightway channels. After being loaded into the two silica carriers by hydrogen bond, crystalline IMC converted to amorphous form, leading to the improved drug dissolution. And IMC loading capacity of A-CMSN was higher than MCM-41 because curled loading process originating from curvature chiral channels can hold more drug molecules.Compared with IMC, IMC loaded A-CMSN presented obviously fast release throughout the in vitro release experiment, while IMC loaded MCM-41 released faster than IMC at the initial 5 h then showed controlled slow release afterwards, which was closely related to the mesoporous silica nanoparticles and different channel mesostructures of these two carriers. A-CMSN possessed nano-cocoon morphology with curled 2D hexagonal channel and its channel length was shorter than MCM-41, therefore IMC molecules can easily get rid of the constraint of A-CMSN then to be surrounded by dissolution medium.
1. Introduction
Chirality is ubiquitous in nature [1] and may be most pronounced in biomolecules such as proteins and DNA [2] . Recently, the synthesis of chiral materials has drawn much attention due to the potential applications of chiral materials in enantioselective catalysis, chiral selectivity separation and chiral recognition [3] , which was a great contribution to the fields of pharmacy, biochemistry and optics devices [4] . Scientists have been devoted to developing its synthesis mechanism [5] , chemistry chirality [6] and specific and advanced functions [7] . Up to now, the synthesis of chiral mesoporous silica has been promoted mainly by template-directing methods such as chiral cationic surfactant-directing method [8] and chiral anionic surfactant directing method [9,10] , resulting in the generation of chiral mesoporous silica from the cooperative self-assembly of amphiphiles and silicates.
Because of its potential applications in drug delivery,adsorption and chiral separation, it was of great interest to have organo-functionalized hybrid chiral mesoporous silica materials on the external and/or internal surfaces. It was generally possible to synthesize organosilane-functionalized chiral mesoporous silica materials by these three methods,co-condensation, post-grafting and imprint coating methods [11,12] . Among the three methods, post-grafting and co-condensation were the two popular ones. The former indicated that the mesoporous silica nanoparticles were first synthesized, and then the surface was modified with functional groups, the latter referred to the silica source and organosilane hydrolyze and condense [13] . Compared with the post-grafting method, the co-condensation method was better because of its ability to make the distribution of organic groups more uniform, higher loading, and did not need to close the advantages of mesopores [14] . As far as we know, amino-functionalized mesoporous silica materials have been reported for many applications in waste-water treatment, base catalysis, absorption for heavy metal ions,enzyme immobilization, further post-synthesis functionalization, and toxic oxygen anions. However, the application of amino-modified chiral mesoporous silica in drug delivery systems has rarely been reported [15-17] .
Much work has been done to improve the solubility of poorly water-soluble drugs, such as nanoparticles, solid dispersion, micronization and co-solvents [18,19] . Among them,nanotechnology has been developed as an alternative method to formulate poorly water-soluble drugs [20-22] . The Noyes-Whitney and Ostwald-Freundlich equations showed that the solubility of poorly water-soluble drugs can be ameliorated by making the particle size become small to nanoscale, thus solving the challenges of the above formulation [23-25] . Unfortunately, the nanoparticle's small particle size resulted in reduced surface free energy and therefore instability, leading to the need for more effective stabilizers or nanoparticle formulation matrices [26] . Due to the unique properties of mesoporous silica nanoparticles: stable properties, large surface area and large pore volume, it was very effective in making up for this disadvantage [13,25] . Therefore, chiral mesoporous silica nanoparticles with ordered mesostructure can be selected to load poorly water-soluble drugs to increase their solubility and thus their bioavailability. In addition, since chiral mesoporous silica nanoparticles had a crimp structure and a chiral characteristic when using chiral mesoporous silica nanoparticles as carriers, it was of great significance to study chiral mesoporous silica nanoparticles as carriers for poorly soluble drugs.
In this paper, we successfully synthesized amino-modified chiral mesoporous silica nanoparticles based on the selfassembly of chiral anionic surfactants and co-condensation of the structural directing agent (3-aminopropyltriethoxysilane,APTES) and an inorganic silicon source (tetraethoxysilane,TEOS). The reasons for choosing indomethacin (IMC) as a model drug were as follows: (1) IMC, an acidic non-steroidal anti-inflammatory drug, can cause gastrointestinal mucosal irritation due to direct contact of free carboxyl groups with the gastrointestinal mucosa, so loading IMC into mesoporous silica nanoparticles was expected to reduce its side effects[27,28] ; (2) carboxyl group of IMC can probably interact with amino group through hydrogen bonding and other interaction forces, which may improve loading capacity of mesoporous silica nanoparticles to IMC. Given the wide range of applications of mesoporous silica nanoparticels as drug carriers and the characteristic of chiral mesoporous silica nanoparticles, we first attempted to load insoluble IMC into aminofunctionalized chiral mesoporous silica nanoparticlesin order to improve its solubility and dissolution rate. Interestingly,the synthesized carriers possessed morphology similar as cocoons, therefore we name it as amino group functionalized chiral mesoporous silica nano-cocoon (A-CMSN). The morphological characteristics, including mesoporous structure,surface area and pore size distribution of A-CMSN were systematically investigated by Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and nitrogen adsorption surface area analyzer. Drug loading was detected by the application of thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). We believe this study will be help in designing better oral delivery systems that use A-CMSN as carrier.
2. Materials and methods
2.1. Materials
Tetraethoxysilane (TEOS) and 3-aminopropyltriethoxysilane(APTES) were bought from Aladdin (Shanghai, China). IMC was purchased from Dalian Meilun Biotechnology Co. Ltd. (Dalian,China. Palmitic acid and L-alanine methyl ester, N, N '- dicyclohexyl carbon imine (DCC) and N-dimethyl aminopyridine(DMAP) were obtained from Chengdu Xiya Chemical Technology Co. Ltd. (Chengdu, China). All other chemicals were reagent grade and deionized water was prepared by ion exchange.
2.2. Synthesis of N-PLA
Solution A was typically prepared by dissolving palmitic acid and DMAP at a 10:1 molar ratio of the complex into 100 ml of dichloromethane, and the mixture was then stirred in a 0 °C water bath for 1 h. 3 ml triethylamine and 26 mmol L-alanine methyl ester were mixed in 50 ml methylene chloride and then stirred at ambient temperature for 40 min to prepare solution B. Thereafter, DCC solution was prepared by dissolving 28.4 mmol DCC in 50 ml methylene chloride. When the preliminary work finished, Solution B was completely added to Solution A, and then the DCC solution was added slowly to the mixed solution. The system was stirred at 0 °C for 3 h in a water bath and then stirred overnight at ambient temperature.The resulting solution was centrifuged and washed sequentially with water, saturated sodium chloride solution, saturated sodium bicarbonate solution, hydrochloric acid (1 M) and again water. The organic layer was dried over magnesium sulfate and filtrated. The organic solvent was removed by evaporation to give the crude product which was purified by crystallization from a mixed solution of ethyl acetate/n-hexane with a volume ratio of 1:1 to obtain a white solid as the final product. 1H NMR (400 MHz, CDCl3) δ 6.00 (d, 1H), 4.58-4.66 (m, 1H),2.21 (t, 2H), 1.60-1.67 (m, 2H), 1.41 (d, 3H), 1.24-1.31 (m, 24H),0.89 (t, 3H).
N-PLA was synthesized by hydrolysis of N-PLA methyl ester. Under a water bath at 0 °C, 3 mmol N-PLA methyl ester was dissolved with a certain amount of methanol. Afterwards, 1 M sodium hydroxide was added to the above mixture, and the methanol remaining in the mixture was distilled off under reduced vacuum. The product was isolated by addition of hydrochloride acid and the pH of the resulting mixture was adjusted to about 2. Finally, the white precipitate was filtered,washed with water and then dried at 60 °C to obtain N-PLA product [29] .1H NMR (400 MHz, CDCl3) δ 6.02 (d, 1H), 4.58 (m,1H), 2.24 (t, 2H), 1.62-1.65 (m, 2H), 1.47 (d, 3H), 1.27 (d, 24H),0.88 (t, 3H). The FTIR and 1H-NMR spectrum were displayed in the supporting information.
2.3. Preparation of A-CMSN
In a typical synthesis scheme, 0.33 g N-PLA was dissolved with 10 ml deionized water, and 10 g sodium hydroxide (0.1 M) together with 10 g hydrochloric acid (0.1 M) was then added to the above solution with stirring and the stirring was continued for 1 h at ambient temperature. Thereafter, a mixed solution consisting of 0.24 ml APTES and 1.57 ml TEOS was added dropwise to the above solution with stirring, allowed to react for 24 h, then centrifuged, washed with water and ethanol, and placed it in an oven at a temperature of 60 °C overnight. The synthesized sample was extracted with ethanolamine (17%,v/v) in ethanol at its boiling point for 12 h. A-CMSN was then obtained by ion exchange between amino groups and amines[30] .
2.4. Characterization of A-CMSN
The critical micelle concentration of N-PLA was determined using a DDS-11A conductivity meter (Shanghai Rex New Rock Instruments Co., Ltd.). The surface morphology of A-CMSN was characterized using a SURA35 field emission scanning electron microscope (ZEISS, Germany). Before examination,the sample was mounted on a metal stub using double-sided tape and sputtered with a thin gold layer under vacuum. The A-CMSN was ultrasonically dispersed in ethanol and then deposited on carbon-coated copper grids before using Tecnai G2 F30 TEM instrument to characterize its pore structure. FTIR(Spectrum 1000, Perkin Elmer, USA) spectra of A-CMSN was recorded from 400 to 4000 cm -1 in transmittance mode with a resolution of 1 cm-1. A-CMSN was milled and mixed with dried KBr in an agate mortar and pestle to prepare KBr disk. ACMSN was placed in an aluminum crucible and heated from 0 to 300 °C with a differential scanning calorimeter (DSC, Q1000,TA Instrument, USA) under a nitrogen flow at a heating rate of 10 °C/min.
2.5. IMC loading into A-CMSN
Solvent deposition, a combination of immersion and solvent evaporation, was used to load IMC into A-CMSN aiming to synthesize a drug delivery system that was uniform and delivered high drug load. Specifically, the IMC was dissolved in acetone to give a concentration of 15 mg/ml, and then 2 ml of this solution was mixed with 90 mg A-CMSN and stirred for 24 h, then dried it in vacuum to evaporate the solvent. It should be noted that MCM-41 with 2D hexagonal mesostructure similar as ACMSN but no curled morphology, was introduced to compare loading capacity and in vitro drug release with A-CMSN, aiming to investigate the potential unique ability of A-CMSN to load and release IMC (carboxylic acid drugs). For the sake of clarity, the drug delivery system of A-CMSN loaded with IMC will be referred to as C, and the drug delivery system of MCM-41 loaded with IMC will be referred to as M.
The actual drug loading of C and M was ascertained using thermal gravimetric analysis (TGA), which was performed from 30 to 600 °C with a TGA-50 instrument (Shimadzu, Japan)at a heating rate of 10 °C/min under a nitrogen flow of 40 ml for 1 min.
2.6. Characterization of IMC loaded A-CMSN
Fourier transform infrared spectroscopy (FTIR, Spectrum 1000,Perkin Elmer, USA) spectra of samples were collected over the spectral region of 400-4000 cm-1. Before examination, samples were mixed with KBr to make small disks for testing.The samples were placed in an aluminum crucible and heated from 0 to 300 °C with a differential scanning calorimeter (DSC,Q1000, TA Instrument, USA) under a nitrogen flow at a heating rate of 10 °C/min.
2.7. In vitro release of IMC from A-CMSN
In vitro dissolution experiments were performed using a USP I basket stirring method (100 rpm, 37 °C and 250 ml dissolution medium) and a ZRD6-B Dissolution Tester (Shanghai Huanghai Pharmaceutical Testing Instrument Factory). The powdery state of C or M was passed through 80 mesh sieve and was filled into the same type of hard gelatin capsules. C and M capsule samples equivalent to 10 mg IMC and 10 mg IMC capsule samples were added to simulated intestinal fluid (pH 6.8). At predetermined time points, 5 ml of sample was removed from the release media, and meanwhile, an equal amount of fresh medium was added to maintain a constant volume of dissolution. Samples that passed through a 0.45 μm microporous
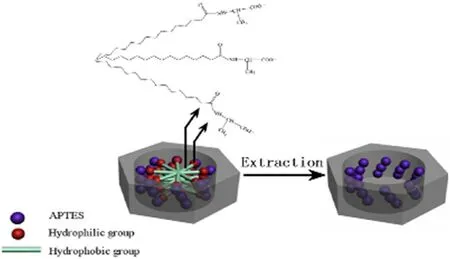
Scheme 1 -Schematic illustration of the template for generating A-CMSN.
membrane were tested at a wavelength of 320 nm using a UV-1750 spectrophotometer (Shimadzu, Japan).
3. Results and discussion
3.1. Nano-cocoon structure of A-CMSN
It was generally accepted that the formation mechanism of chiral mesoporous silica nanoparticles belonged to cooperative self-assembly process in which the charge density of the soluble silica source initially determines the interfacial packing density of the surfactant and the biphasic geometry of the surfactant shape. This can be coordinated to a given direction of the soluble silica source and surfactant which was advantageous relative to the source of soluble silica source [31] . In our synthesis of A-CMSN, anionic surfactant (N-PLA), co-structure directing agent (APTES) and silica source (TEOS) were applied to construct the pattern of S-M + I-, in which S-, M + and I-represent an anionic surfactant, a co-structure directing agent with a cationic amino group and soluble silica source with a negative charge, respectively. Herein, we described in detail the formation process of A-CMSN as outlined in Scheme 1 . Since the concentration of N-PLA (0.337 mol/l) was much higher than the critical micelle concentration of N-PLA(0.001 mol/l, measured by using conductivity meter) before adding the mixed solution of APTES and TEOS, an internally assembled hydrophobic group and an externally extended hydrophilic group were formed micelles. In addition, due to the balance between carboxyl groups and carboxylates, a large amount of negatively charged carboxylate of N-PLA was present in the alkaline aqueous solution (pH = 8). After adding APTES and TEOS into the above solution, the negative charged head group of N-PLA interacted with the positively charged ammonium sites of the APTES [4] . In this self-assemble process, the negative charge density in alkaline aqueous solution was high, so the electrostatic repulsion caused by the hydrophilic groups was quite strong, resulting in higher curvatures of the amphiphilic molecules and staggered packing [32] to reduce the surface free energy [33] . Mesoporous silica nanoparticles with a crimped mesoporous structure were finally formed. Simultaneously, the alkoxysilane site of APTES polymerized with the alkoxysilane site of TEOS to promote silicate organization around the amphiphilic micellar superstructure. Meanwhile, silicate polymerization also promoted the aggregation of amphiphiles to form an ordered liquid crystalline intermediate structure p6mm [5] .
A-CMSN had a typical two-dimensional hexagonal p6mm chiral mesostructure with ordered hexagonal mesopores( Fig. 1 A, a) viewed downwardly or upwardly from the schematic of A-CMSN ( Scheme 1 ), meanwhile channels with curvature were obtained from lateral direction. After amplifying A-CMSN and MCM-41 to the same magnification, we confirmed that A-CMSN with a 2D hexagonal cocoon-like morphology possessed hexagonal ordered channels, whose curvature was clearly observable, which was quite different from MCM-41 with straightway 2D hexagonal channels. Moreover,there is no significant difference in particle size and channel diameter (about 6.25 nm) between A-CMSN and MCM-41,demonstrating that the channel curvature is a major contributor to the discrimination of these two mesoporous silica nanoparticles. Therefore, it is interesting to see how the curled channels within the nanometer range affect drug loading and release behavior.
3.2. Characteristics of IMC loaded A-CMSN
In order to further demonstrate that IMC were effectively incorporated into A-CMSN, and also to investigate their possible interactions, the FTIR analysis of IMC, MCM-41, IMC loaded MCM-41 (M), A-CMSN, and IMC loaded A-CMSN (C) were carried out and their spectrums were presented in Fig. 2 . Pure IMC showed its characteristic peaks at 1718 and 1692 cm-1, which belonged to the carbonyl groups of the acid and amide [34] ,and characteristic peak appeared at 1308 cm -1 was assigned to aromatic C -N stretching vibration. Comparing A-CMSN with MCM-41, a new band belonged to NH 2 bending was detected at 1464 cm -1 and two bands attributed to CH2stretching were observed at 2919 and 2850 cm -1 due to the introduction of the amino propyl group from APTES [35] . When IMC was encapsulated into MCM-41, the carbonyl peaks were not observed, whereas a slight shift in the aromatic C -N stretching vibration was observed, indicating that the carbonyl group in the IMC molecule were involved in the formation of the hydrogen bond with the silanol groups of MCM-41. After IMC was loaded into A-CMSN, it was clearly observed that the carbonyl peaks disappeared, indicating that the carboxylic acid in the IMC molecule participated the formation of the hydrogen bond with the amino groups functionalized at the internal surface of A-CMSN. In the meanwhile, the carbonyl group adjacent to the amino group formed a hydrogen bond with the silanol groups on the surface of the silica, and this hydrogen bond also proved that the peak belonging to the aromatic C -N stretching vibration at 1318 cm -1 showed a sharp decrease.

Fig. 1 -TEM images of (A) A-CMSN scanned in up to down or down to up direction (a) and in lateral direction (b); (B) A-CMSN and MCM-41 scanned in lateral direction.
To evaluate the physical state of IMC in A-CMSN, DSC measurement of IMC, MCM-41, IMC loaded MCM-41 (M), A-CMSN and IMC loaded A-CMSN (C) were performed and the results were shown in Fig. 3 . As displayed in Fig. 3 , the IMC thermogram had a single sharp endothermic peak at 163.5 °C, which was consistent with the melting point of crystalline IMC. In the case of MCM-41 and IMC loaded MCM-41, no depression was observed, illustrating that the absence of phase transitions of IMC, demonstrating that the IMC was in an amorphous state upon loading into MCM-41 [36] . From the thermogram of A-CMSN, endothermic phenomenon was clearly observed as a result of the decomposition of aminopropyl groups during the heat treatment. However, the thermogram of IMC loaded into A-CMSN did not show any characteristic endotherm peaks belonging to IMC, which provided indirect evidence that IMC was fully contained in A-CMSN. It was speculated that the limited mesoporous structure and the curled mesostructure of A-CMSN prevented the IMC from crystallization due to space constraint, and the hydrogen bonds formed between the IMC and the carries might also be related to the amorphous state of IMC [37,38] .
In the procedure of IMC loading, TGA was used to quantify drug adsorption capacity of IMC loaded A-CMSN ( Fig. 4 C)and IMC-loaded MCM-41 ( Fig. 4 M). The weight loss of pure IMC ( Fig. 4 I) began to decrease at about 200 °C and gradually reached 100% at 600 °C, indicating that the determination of the temperature range was appropriate. For A-CMSN, an initial weight loss of 8.54% can be observed between 50 and 200 °C due to aminopropyl decomposition, and at a temperature range of 200-600 °C, the weight loss of IMC loaded ACMSN was measured to be 31.82%. It was clearly observed that the weight loss of IMC adsorbed into A-CMSN was higher than that of MCM-41 (23.65%), and explanations can be given as follows: (1) the hydrogen bonding force formed between IMC and A-CMSN was stronger than that of MCM-41, which was because that in addition to silanol groups, the amino groups of APTES can also bind to the carboxyl groups of IMC [27] ; (2) after amino-functionalization, the electrostatic or hydrophobic interactions can be introduced leading to a higher degree of IMC loading [13] ; (3) because A-CMSN had a two-dimensional curled hexagonal channel nano-cocoon, IMC molecules had more chances to be loaded into A-CMSN compared to MCM-41 possessing straightway 2D hexagonal channel, and the schematic diagram can be seen in the small image inserted in Fig. 4 .
3.3. IMC release from A-CMSN
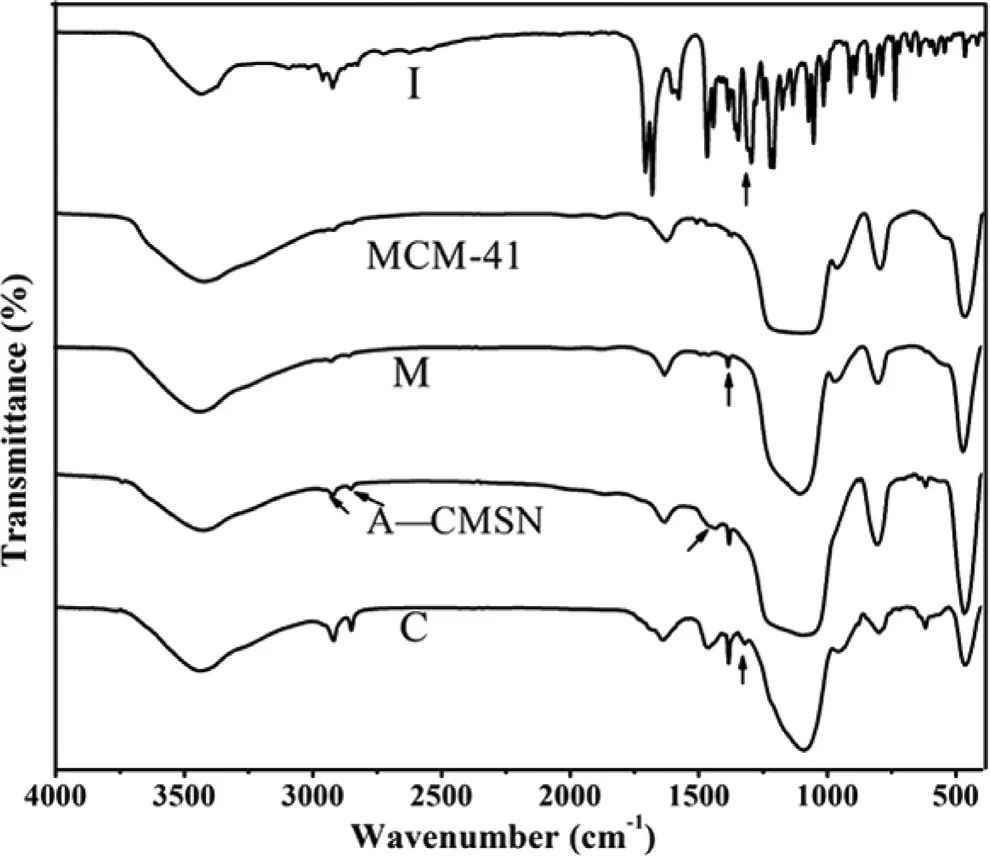
Fig. 2 -FTIR spectra of IMC (I), MCM-41, IMC loaded MCM-41 (M), A-CMSN, IMC loaded A-CMSN (C).
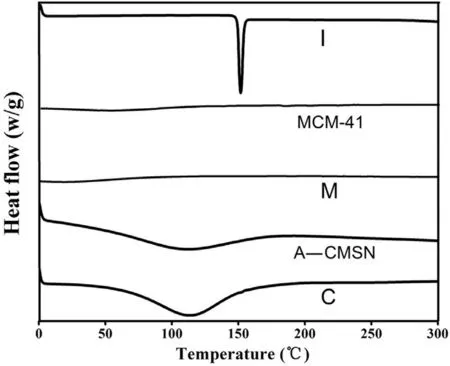
Fig. 3 -DSC thermograms of IMC (I), MCM-41, IMC loaded MCM-41 (M), A-CMSN, IMC loaded A-CMSN (C).
It can be concluded that the two mesoporous silica nanoparticles, A-CMSN and MCM-41, had unique functions as IMC carriers based on the result of in vitro release. In contrast with pure IMC ( Fig. 5 I), IMC loaded A-CMSN ( Fig. 5 C) presented a significant rapid release throughout the in vitro process, whereas IMC loaded MCM-41( Fig. 5 M) released faster than pure IMC at the initial 5 h and then controlled slow release, which was closely related to the mesoporous silica nanoparticles and different channel mesostructures of these two carriers. It was well-known that making the drug into an amorphous state and reducing its particle size can improve the drug solubility and dissolution rate. Nanoparticles or nanotechnologies have long been considered as effective methods to improve the dissolution rate because nanocrystals increase the area for dissolution due to their small particle size. In addition, IMC can only exist in an amorphous state due to the pore size limitations[39] . Therefore, after IMC was loaded into A-CMSN and MCM-41, the results of DSC analysis showed that IMC was present in an amorphous state, resulting in an increased apparent solubility and dissolution rate of IMC. It was noteworthy that IMC loaded MCM-41 displayed a special case, its controlled slow release effect after 5 h, and this effect was correlated to its channel mesostructure. Since MCM-41 contained straightway 2D hexagonal channel, the dissolution medium can hardly induce the release of IMC molecules located at the relative innermost sites of MCM-41, resulting in the sustained release effect.On the contrary, A-CMSN possesses nano-cocoon morphology with curled 2D hexagonal channel and its channel length was shorter than MCM-41, therefore IMC molecules can easily get rid of the constraint of A-CMSN then to be surrounded by dissolution medium (whose schematic diagram was shown in Fig. 5 ).
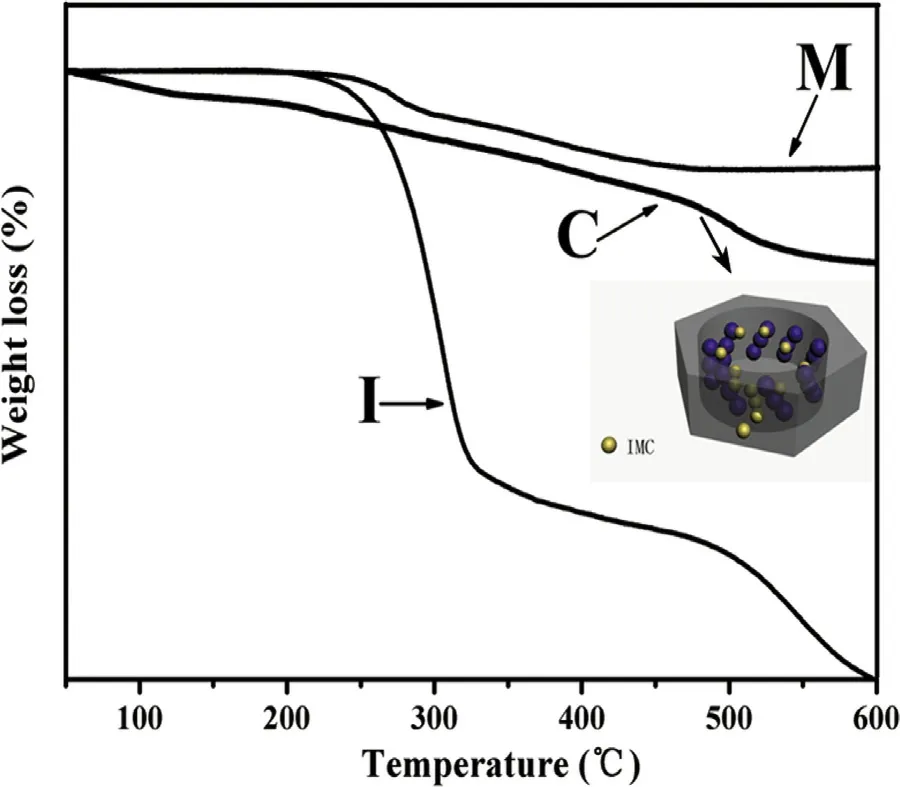
Fig. 4 -TGA curves of IMC (I), IMC loaded MCM-41 (M), and IMC loaded A-CMSN (C). Schematic image of IMC loading into A-CMSN was inserted.
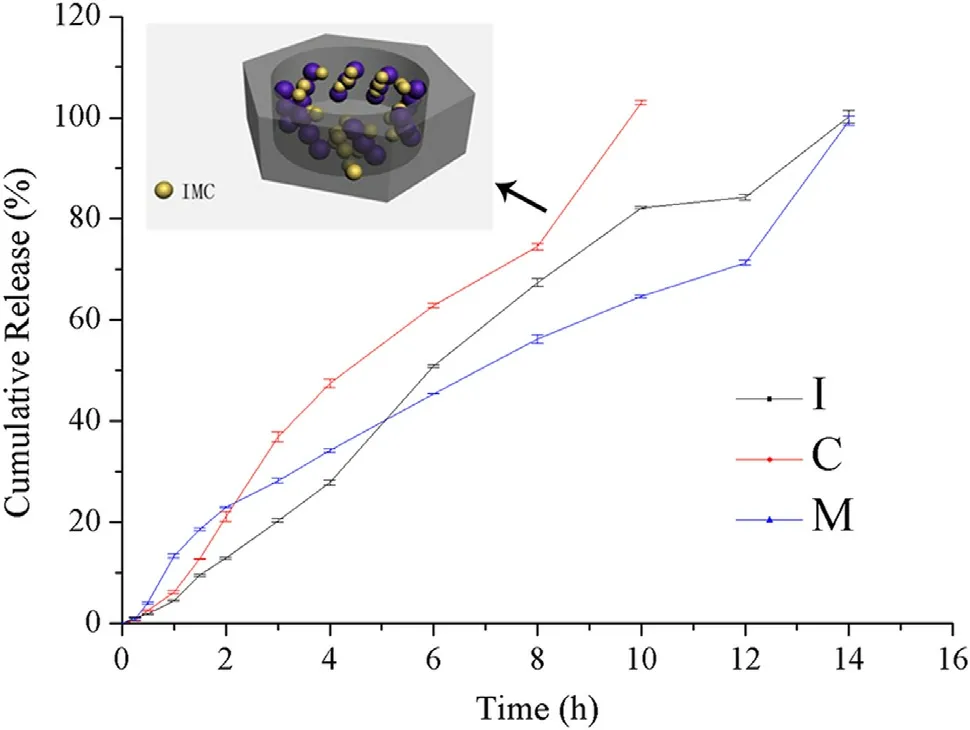
Fig. 5 -Release profiles of IMC (I), IMC loaded MCM-41 (M),and IMC loaded A-CMSN (C). Schematic image of IMC releasing from A-CMSN was inserted.
4. Conclusion
A-CMSN was synthesized using co-condensation method based on self-assembly of chiral anionic surfactant as a template. A-CMSN had two-dimensional hexagonal nano-cocoon morphology due to the curvature channel formed by chiral anionic surfactant, which was quite different from MCM-41 with straightway channels. Crystalline IMC converted to amorphous form after being loaded into two silica carriers by hydrogen bonds, leading to the improved drug dissolution.A-CMSN had a slightly higher loading capacity to IMC than MCM-41 because when using A-CMSN as a silica support, the curled channel accommodated more drug molecules. Compared with pure IMC, IMC loaded A-CMSN presented a significant rapid release throughout the in vitro process, whereas IMC loaded MCM-41 released faster than pure IMC at the initial 5 h and then controlled slow release, which was closely related to the mesoporous silica nanoparticles and different channel mesostructures of these two carriers. In this paper,we studied the synthesis of chiral mesoporous silica nanoparticles with curled channels and their application in drug loading and release of IMC, which may help to design a unique chiral mesoporous silica material and its application in the pharmaceutical field.
Conflicts of interest
No conflict of interest exits in the manuscript, and the article was approved by all authors.
Acknowledgments
This work was supported by Postdoctoral Science Foundation of China 2017M611268 .
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ajps.2018.04.002 .
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- The appliances and prospects of aurum nanomaterials in biodiagnostics, imaging, drug delivery and combination therapy
- Current developments in drug delivery with thermosensitive liposomes
- The application of nitric oxide delivery in nanoparticle-based tumor targeting drug delivery and treatment
- Delivery of docetaxel using pH-sensitive liposomes based on D - α-tocopheryl poly(2-ethyl-2-oxazoline)succinate: Comparison with PEGylated liposomes
- Optimizing pH-sensitive and time-dependent polymer formula of colonic pH-responsive pellets to achieve precise drug release
- A hybrid genipin-crosslinked dual-sensitive hydrogel/nanostructured lipid carrier ocular drug delivery platform