越南水稻黄矮病毒多克隆血清抗体的制备
2019-09-10何越强陈氏懦花陈阮河杜新勇阮德辉韦善富覃武吕荣华
何越强 陈氏懦花 陈阮河 杜新勇 阮德辉 韦善富 覃武 吕荣华




摘要:【目的】制備用于检测水稻黄矮病毒(RYSV)的多克隆抗体,为水稻和黑尾叶蝉上的病毒诊断提供技术支持。【方法】利用两种不同来源的RYSV抗原免疫家兔,一种是从受感染的水稻病叶组织纯化获得RYSV病毒粒子;另一种是从越南RYSV分离株克隆出完整N蛋白编码基因,然后将其连接至pET-28a载体上,在大肠杆菌Rosetta(DE3)菌株中进行诱导表达获得N蛋白抗原。其中,N蛋白抗原又以两种形式(洗脱纯化N蛋白和N蛋白条带聚丙烯酰胺切片匀浆)对家兔进行免疫。最后,采用PTA-ELISA分析评估家兔抗体血清(多克隆抗体)对水稻叶片和黑尾叶蝉RYSV的特异性和敏感性。【结果】分离自越南RYSV分离株的N基因由1542个核苷酸组成,与来自我国和日本RYS分离株N基因序列进行比对,其核苷酸序列同源性分别为98.1%和97.9%,对应的推导氨基酸序列同源性为83.4%和99.4%。以RYSV病毒粒子、洗脱纯化N蛋白和N蛋白条带聚丙烯酰胺切片匀浆3种抗原免疫家兔获得的多克隆抗体均能有效检测出水稻植株中的RYSV,其中,感病植株的OD405分别为1.449、 2.337和1.649,健康植株的OD405分别为0.375、0.294和0.283。PTA-ELISA检测结果表明,获得的多克隆抗体能从单个带病毒黑尾叶蝉中检测出RYSV,且该结论在RT-PCR检测中得到进一步验证。【结论】制备获得的多克隆抗体对RYSV具有较高特异性和敏感性,可用于水稻和黑尾叶蝉上的病毒诊断,同时表明以含有病毒植物蛋白的聚丙烯酰胺切片直接注射免疫模型动物制备多克隆抗体具有可行性。
关键词: 水稻黄矮病毒; 多克隆抗体; 酶联免疫吸附测定(ELISA); N蛋白; 表达; 病毒纯化
0 Introduction
【Research significance】Yellow stunt disease of rice caused by rice yellow stunt virus(RYSV) [syn. rice transitory yellowing virus(RTYV)] (Hiraguri et al.,2010) was firstly observed in Guangxi of China in 1957 and subsequently,numerous outbreaks of the disease have been reported in 1960s and 1970s in southern and central China,Taiwan(China),Japan,Thailand and Vietnam(Hibino,1996). Recently,the yellow stunt disease of rice has reoccurred in several provinces of Vietnam,particularly in summer(Ha et al.,2010). 【Research progress】RYSV is member of the genus Nucleorhadovirus of the family Rhadoviridae. Like other rhabdoviruses,RYSV has enveloped and bullet- or bacilliform-shaped virions,and monopartite,negative-sense,single-stranded RNA(ssRNA) genome that is 12-14 kb in length(Dietzgen et al.,2011;Hiraguri et al.,2010). The genome of RYSV is cha-racterized by a 3'-leader,a 5'-trailer,and seven open reading frame(ORF) in the order 3'-N-P-3-M-G-6-L-5',which encodes for seven proteins,namely nucleocapsid protein(N),phosphoprotein(P),P3 protein,matrix protein(M),glycoprotein(G),P6 protein and polymerase(L)(Fang et al.,1994; Huang et al.,2003; Luo et al.,1998; Luo and Fang,1998; Wang et al.,1999; Hiraguri et al.,2010). RYSV is naturally transmitted by three species of rice green leafhoppers,Nephotettix apicalis,N. cincticeps and N. impicticeps in a propagative persistent manner but not transmitted via eggs(Hibino,1996; Jackson et al.,2005). Just far,rice is the only known host of RYSV. Typical symptoms on rice are leaf yellowing,reduced tillering,and mild stunting. The infected plants may recover and produce symptomless leaves. In infected rice plants,RYSV is limited in the phloem tissues(Ou,1985;Hibino,1996).【Research breakthrough point】Due to yellowing appearance of the infected plants,diagnosis of the disease is difficult. Furthermore,the detection of the virus in the vectors is impossible without help of diagnostic tools. Because the genome of RYSV is RNA,the virus can be detected by methods such as traditional RT-PCR(Ha et al.,2010) or reverse-transcription loop-mediated isothermal amplification(RT-LAMP)(Le et al.,2010). Ho-wever,these methods require expensive facilities and reagents and is relatively time-consuming. Currently,the most popular tool for diagnosis of plant viruses is enzyme linked immune sorbent assay(ELISA) that rely on the availability of specific antibodies or antisera. Compared with the RNA-based methods,the ELISA is much cheaper and easier,particularly when a large number of samples are tested(Uehara-Ichiki,et al.,2013). Prior to this study,only one antibody specific to virion of RYSV was produced in Japan and demonstrated to be useful in detection of RYSV in both rice plants and insect vectors(Takahashi et al.,1988),Unfortunately,it is not able to obtain this antibody for diagnosis of RYSV in Vietnam. 【Solving problems】Therefore,the major aim of this work is to produce antibodies specific to RYSV for diagnosis of RYSV in Vietnam. In this study,it was demons-trated that both purified virions and N protein of RYSV expressed in E. coli cells could induce formation of antibodies specific to this virus in the immunized rabbits.
1 Materials and methods
1. 1 Experimental materials
Rice green leaf hoppers and infected rice plants showing typical symptoms were collected from su-mmer crops in outbreak regions of Bacgiang Province in 2013 and 2014. The collected samples were kept in net-house of the Research Center of Tropical Plant Diseases of Vietnam National University of Agriculture for further research.
1. 2 Reverse Transcriptase(RT-PCR),cloning and sequencing
Primers for the diagnosis of RYSV or expre-ssion of its N protein were presented in Table 1. Total RNA was extracted from the leaf tissues by using a RNeasy Plant Mini Kit(Qiagen) following the manufacturer’s instructions.
The complete open reading frame(ORF) of the N protein was amplified by a two-step RT-PCR procedure. cDNA was synthesized using the RY-left-F1 primer and a ReverAid Premium Reverse Transcriptase Kit(Thermo Scientific). Then,cDNA was subjected for PCR using a Gotaq Green Master Mix (Promega) and two primers,RY-N-F and RY-N-R. The PCR conditions were as follows: initiation denaturation at 94 ℃ for 4 min,then 25 cycles of denaturation 94 ℃ for 30 s,annealing at 55 ℃ for 45 s and extension at 72 ℃ for 1 minute 30 seconds,and final elongation at 72 ℃ for 5 min. RT-PCR produ-cts were purified from agarose gel using a GeneJet Gel Extraction Kit(Thermo Scientific). Two ends of the gel purified amplicons were treated with BamH I and Sac I and the amplicons were further purified using a GeneJet PCR Purification Kit(Thermo Scientific). The treated-end amplicons were ligated into the plasmid pET-28a(Novagen) which was opened by BamH I and Sac I double digestion. The recombinant plasmid was transformed into the XL1-Blue competent cells of Escherichia coli(Stratagene) for cloning and sequencing. The positive purified plasmid was sequenced at Macrogen,Inc. (South Korea). The positive plasmid was then transformed into the BL21(DE3) and Rosseta(DE3) competent cells of E. coli(Novagen) for expression. All bacterial transformations were done by using a heat shock method(Inoue et al.,1990). Recombinant colonies were selected on Luria-Bertani(LB) agar medium supplemented with 12.5 μg/mL tetracyclin and 50 μg/mL kanamycin for XL1-blue,50 μg/mL kanamycin for BL21(DE3),34 μg/mL chloramphenicol and 50 μg/mL kanamycin for Rosetta(DE3). The clones were verified by PCR using T7-Pro and T7-Ter primers.
1. 3 Expression and purification of N protein
To test the expression of the N protein,a single fresh bacterial colony bearing expression plasmid was grown at 37 ℃ with shaking at 220 r/min in 2 mL LB medium supplemented with 50 μg/mL kanamycin for BL21(DE3),34 μg/mL chloramphenicol and 50 μg/mL kanamycin for Rosetta(DE3) at OD600 of 0.6 then,50 µL was inoculated in 5 mL of fresh LB medium supplemented with antibiotics at the same concentrations. The bacterium was grown at 25 ℃ with shaking at 220 r/min until with an OD600 of 0.6. At this stage,expression of N protein was induced by addition of isopropyl β-D-1-thiogalactopyranoside(IPTG,Thermo Scientific) to a final concentration of 1 mmol/L. The culture was allowed to grow for two more hours. The cells were harvested by centrifugation at 5000×g for 15 min,and resuspended in 100 µL lysis buffer(20 mmol/L Tris-HCl,2.5 mmol/L EDTA,5 mmol/L imidazole,pH 8.0). The cell suspensions and supernatants were stored at -20 ℃ for further analysis.
The recombinant fusion protein containing 6×His residues at the N-terminus was purified using nickel affinity chromatography(Ni-NTA-agarose) under native conditions. The cells from 700 mL culture of a positive clone cultured and induced at the same conditions were harvested and resuspended in 14 mL lysis buffer. The cell suspension was added with lysozyme from chicken egg white(Sigma) to a final concentration of 1 mg/mL and incubated on ice for one hour. Then,the cells were further disrupted by tho-rough homogenization with a Dounce homogenizer (the tube was immersed in the ice water during homogenization). The homogenate was added with Triton X-100(1%),RNase A(1 μg/mL) and DNase I (1 μg/mL),and incubated at 4 ℃ for 15 min. The lysate viscosity was reduced by passing the lysate through a syringe with 18-gauge needle.
The lysate was transferred to a column contai-ning 2 mL slurry of Ni-NTA-agarose beads(Sigma) which was already equilibrated with lysis buffer. The mixture was mixed well and incubated at room temperature for 30 min. The buffer in the mixture was drained and the beads were washed three times with wash buffer(50 mmol/L NaH2PO4,300 mmol/L NaCl,20 mmol/L imidazole,pH 8.0). The recombinant protein was collected with 5 mL of the elution buffer(50 mmol/L NaH2PO4,300 mmol/L NaCl,250 mmol/L imidazole,pH 8.0) in 5 one-mL fractions. The eluted fractions were further dialyzed with PBS buffer before immunization. The fractions of original lysate,ran-off lysate and ran-off washing solution were also collected for analysis.
Expressed protein was analyzed by 13% SDS-polyacrylamide gel electrophoresis(SDS-PAGE) in a Miniprotean III system(BIORAD). Protein bands were visualised by Coomassie brilliant blue staining.
1. 4 Virus purification
Virions were purified from leaves using a procedure of Lockhart et al.(1985). Fresh infected leaf tissue(100 g) was ground in a blender in four vo-lumes of cold extraction buffer(0.1 mol/L sodium citrate,pH 6.5 containing 10% sucrose,0.2% Na2SO3 and 4% activated charcoal). The extract was cloth filtered,acidified to pH 0.5 with glacial acetic acid,stored at 4 ℃ for 4 h,and clarified at 17400×g for 20 min in a Beckman Algera 64G centrifuge. The virus was concentrated by ultracentrifugation at 91500×g for 30 min in a Beckman Optima L-90K ultracentrifuge. The pellet was resuspended in 0.01 mol/L phosphate buffer,pH 7.2 containing 10% sucrose and 0.85% NaCl. The suspension was layered onto a continuous 12%-50% sucrose gradient prepared in 0.01 mol/L phosphate buffer,pH 7.2 containing 0.85% NaCl. The gradient was centrifuged at 107000×g for 45 minutes in a Beckman SW 41 Ti Rotor. The fractions were collected by a medical syringe and verified by RT-PCR. The positive fractions were mixed together and the virus was reconcentrated by centrifu-gation at 139000×g for 30 min. The virus pellet was finally resuspended in 1 mL of 0.01 mol/L phosphate buffer,pH 7.2 containing 0.85% NaCl and stored at -20 ℃ for immunization. The presence of virions in purified preparation was examined using a JEOL 1010 electron microscope at the Institute of Hygiene and Epidemiology(Hanoi).
1. 5 Immunization
The purified virus,the eluted fraction of purified N protein and the homogenate of the slices of polyacrylamide gel containing the N protein band (0.5 mL each) was emulsified with Freund’s complete adjuvant(0.5 mL) for immunization of a rabbit weighting 2.5-3.0 kg by hind leg intramuscular injections. One week after the first injection,the rabbits were further immunized by three one-week interval injections following the similar procedure; except that the Freund’s complete adjuvant was replaced with the Freund’s incomplete adjuvant. The blood(approximately 0.5 mL) was collected from the ear vein of the immunized rabbits at two and four weeks after the first injection for detecting the presence of the anti-RYSV antibodies in antisera. At five weeks after the first injection,the rabbits were killed and the bloods were collected. The bloods were centrifuged at 5000×g to remove blood cells. The antisera were stored in 50%(v/v) glycerol and 0.05%(w/v) sodium azide.
1. 6 Remove of none specific antibodies from antisera
A simple technique adopted from a procedure described previously(Rybicki et al.,1990) was used to remove none specific antibodies from crude polyclonal antisera. The technique comprises three steps.
Preparation of rice protein. Fresh stem and leaf tissues of the healthy plants(200 g) were homogenized in 400 mL of extract buffer(50 mmol/L Tris-HCl,pH 7.5,0.1 mmol/L EDTA,1 mmol/L β-mercaptoethanol). The homogenate was filtered by cheesecloth. The filterate was centrifuged at 5000×g at 4 ℃ for 10 minutes. The total protein in the supernatant was precipitated with(NH4)2SO4 at the final concentration of 80% saturation. The suspension was left overnight at 4 ℃ and centrifuged at 12000×g at 4 ℃ for 15 min to pellet proteins. The protein pellet was then resuspended in 5 mL water and dialyzed in the cold room against 3 times,1-L changes of the PBS buffer overnight to remove residual(NH4)2SO4.
Coating rice protein onto membrane. Nitrocellulose membrane(0.45 μm pore,Sigma) was cut into round sheet with a diameter of approximately 8 cm which fit into a 9 cm Petri disk. The water pre-equi-librated membrane was incubated with the rice protein suspension which was prepared by resuspending the stock protein in PBS buffer at ratio of 1∶10(v/v) in a Petri disk. The disk was incubated overnight at room temperature with gentle shaking. After three washes in PBS-T,the membrane was blocked with 5% skim milk in PBS-T at room temperature for 2 h,followed by three washes in PBS-T.
Removing the non-specific antibodies in antiserum. Original antiserum was diluted 10 times in PBS buffer and was incubated with the coated nitrocellulose membrane in a Petri disk overnight at 4 ℃ with gentle shaking. After incubation,the membrane which absorbed non-specific antibodies was discar-ded and antiserum was kept for further study.
1. 7 Purification of polyclonal antibody
Polyclonal antibody from rabbit antiserum was purified using a Protein A Antibody Purification Kit (PURE1A,Sigma) following the provider’s guideline.
1. 8 PAT-ELISA
A plate-trapped antigen ELISA(PTA-ELISA) format(Mowat and Dawson,1987) was used for all experiments. Test samples(plant or vector) were extracted in carbonate buffer(pH 9.6). One hundred microliters of the extracts were added to the ELISA plate and incubated at 4 ℃ overnight. Then the plate was washed three times with phosphate-buffered saline(pH 7.4) containing 0.05% Tween-20(PBS-T) and blocked with 2% skim milk in PBS-T at room temperature for 45 min. After three washes in PBS-T,antiserum or antibody was diluted in healthy extract that was prepared by extracting fresh healthy leaf tissues in conjugate buffer(PBS-T containing 2%(w/v) polyvinylpyrrolidone(molecular weight 44000) and 0.2%(w/v) ovalbumin(PBS-TPO),added to the plates and incubated at 37 ℃ for 2 h,followed by three washes in PBS-T. Mouse monoclonal anti-rabbit IgG-Alkaline phosphatase(Sigma),diluted 1∶5000 with conjugate buffer was added and incubated at 37 ℃ for 2 h and,finally after three washes,the substrate[4-nitrophenyl phosphate(pNPP) disodium salt diluted at 0.5 mg/mL in 10% diethanolamine buffer,pH 9.8] was added and absorbance was measured at 405 nm after incubation of 60 min. Stop reaction was done by adding 50 µL of 0.5 mol/L NaOH.
1. 9 Sequence analysis
The amplicons were sent for sequencing at the Macrogen Inc.(Republic of Korea). The sequences were initially compared to known viral sequences using the BLAST program available at the National Centre for Biotechnology Information(NCBI)(http://blast.ncbi.nlm.nih.gov/Blast.cgi). Sequences were aligned with the ClustalX program(Larkin et al.,2007). Phylogenetic trees were constructed from the ClustalX-aligned sequences using a MEGA 6.0 software(Tamura et al.,2013).
2 Results and analysis
2. 1 Purification of virus
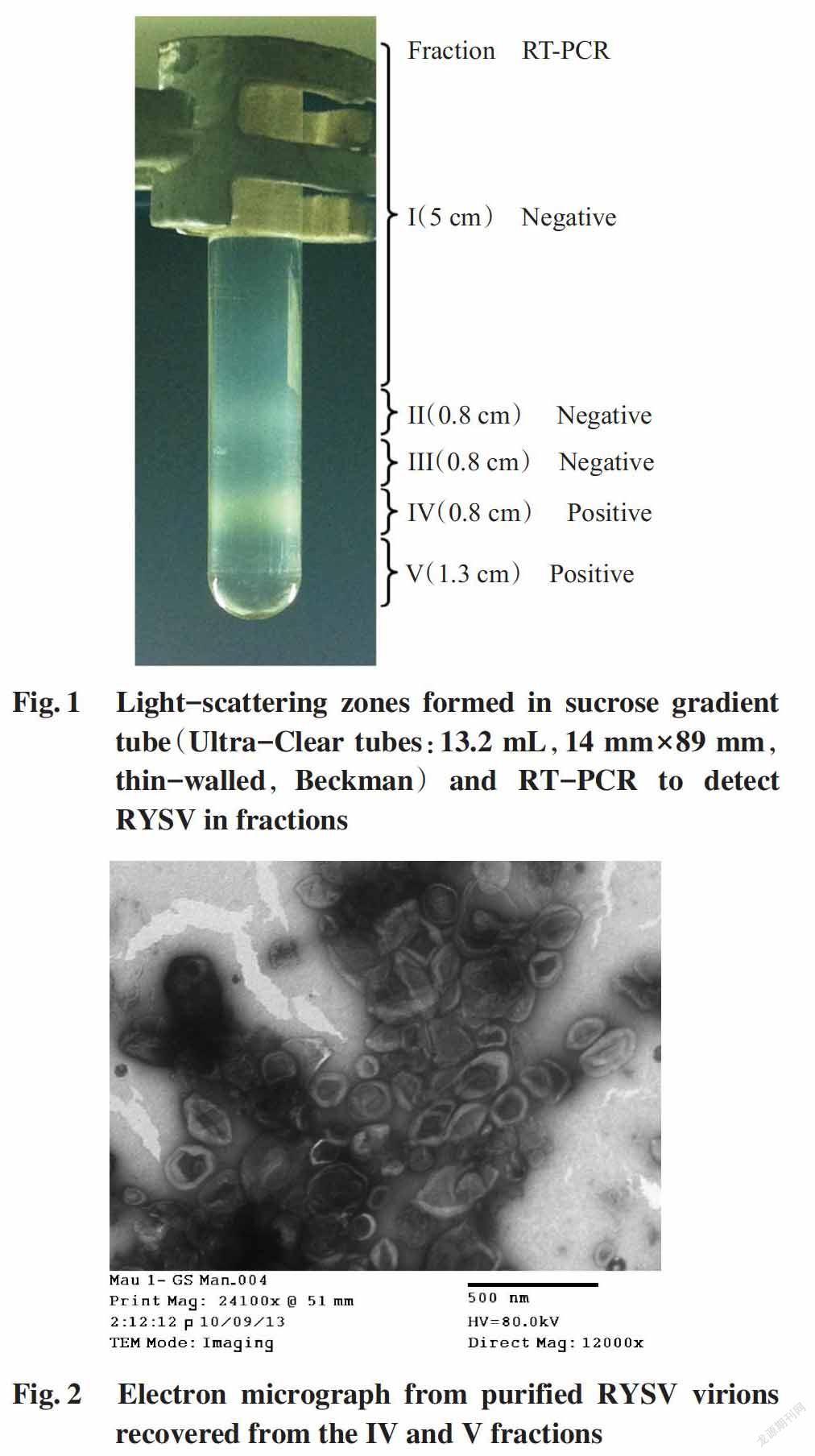
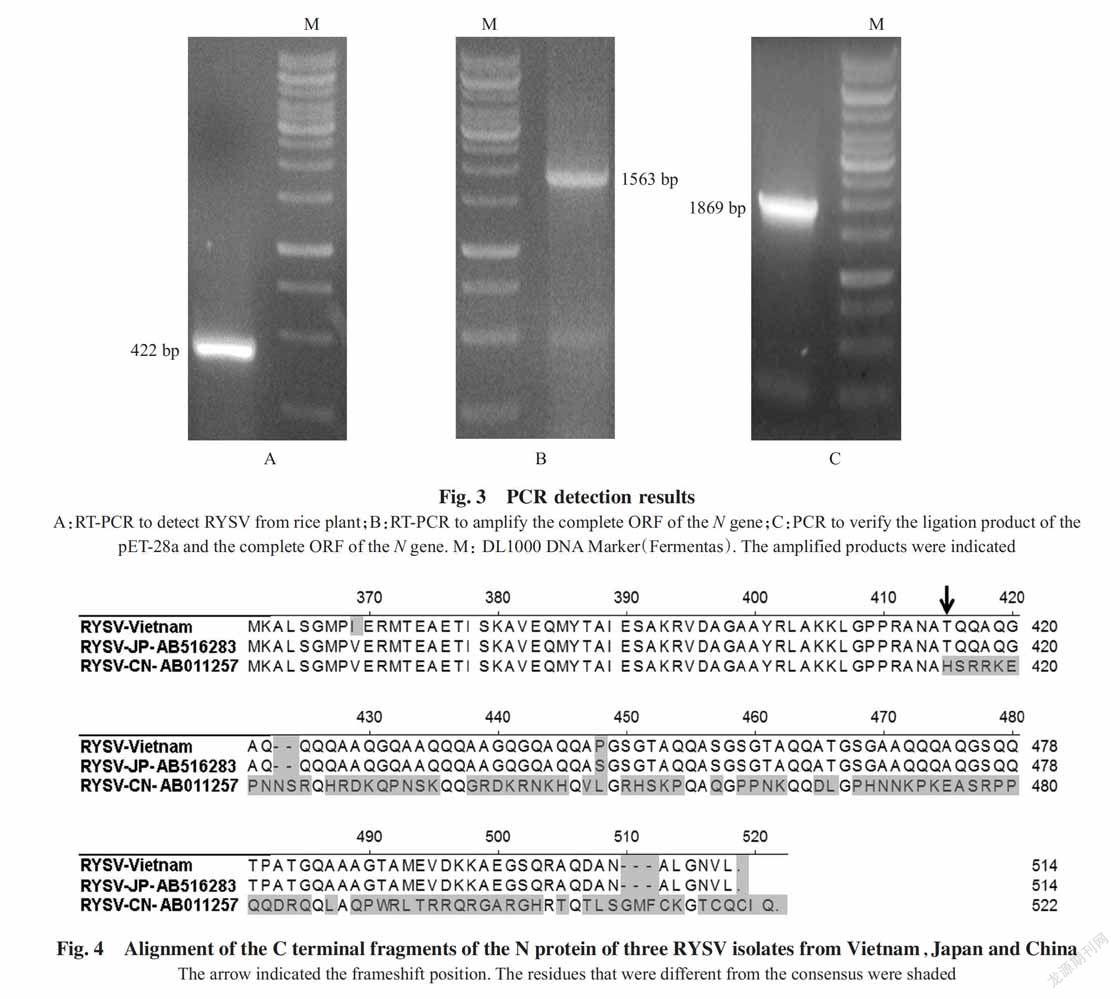
After purification,two well-defined light-scatte-ring opalescent zones were observed in the sucrose gradient tube,and as such,five fragments were formed in the tube(Fig.1). All the fragments were collected,including the pellet that was included in the fragment V. RT-PCR using RY-L-F2 and RY-L-R2 primers were positive for the fragments IV and V(Fig.1),indicating these two fragments contained the virions. The fragments IV and V were then mixed together and the mixture was subjected for a last ultracentrifugation step to pellet the virus. Verification of the pellet preparation under electron microscope revealed the presence of surrounding envelo-ped viral nucleocapsis particles with no internal striations(Fig.2).
2. 2 Cloning and sequencing of the N gene
Total RNA was extracted from a rice plant with typical symptom collected from Bac Giang Province. The RT-PCR using diagnostic primers,RY-L-F2 and RY-L-R2,and the cDNA synthesized with the RY-left-F1 primer as template produced an expected band of 422 bp(Fig.3-A). Using the same cDNA template,the complete ORF of the N gene was PCR amplified with the RY-N-F and RY-N-R primers,which produced a product with expected size of 1563 bp(Fig.3-B). After verifying the ligation reaction by PCR which produced a product with expected size of 1869 bp(1563 bp of the N gene and 306 bp of the vector)(Fig.3-C),the recombinant plasmid(pET-28a contai-ning amplicon) was transformed into XL1-blue cells for cloning. One positive clone,named as pET28-N4 was selected for sequencing.
The sequence of the pET-N4 amplicon was 1567 nucleotide. When compared with the N gene two RYSV isolates available in the GenBank from China and Japan(accession numbers AB011257 and AB516283,respectively),the amplicon had one additional nucleotide at position 1241,which resulted in a frameshift and a termination codon change. Consequently,the N gene of RYSV from Vietnam was composed 1542 nucleotides which was as the same length with that from Japan but 67 nucleotides shor-ter than that from China; and,the sequence of the C terminal fragment of the N protein of RYSV from Vietnam and Japan(99 amino acids in length) was distinct to that from China(107 amino acids in length)(Fig.4).
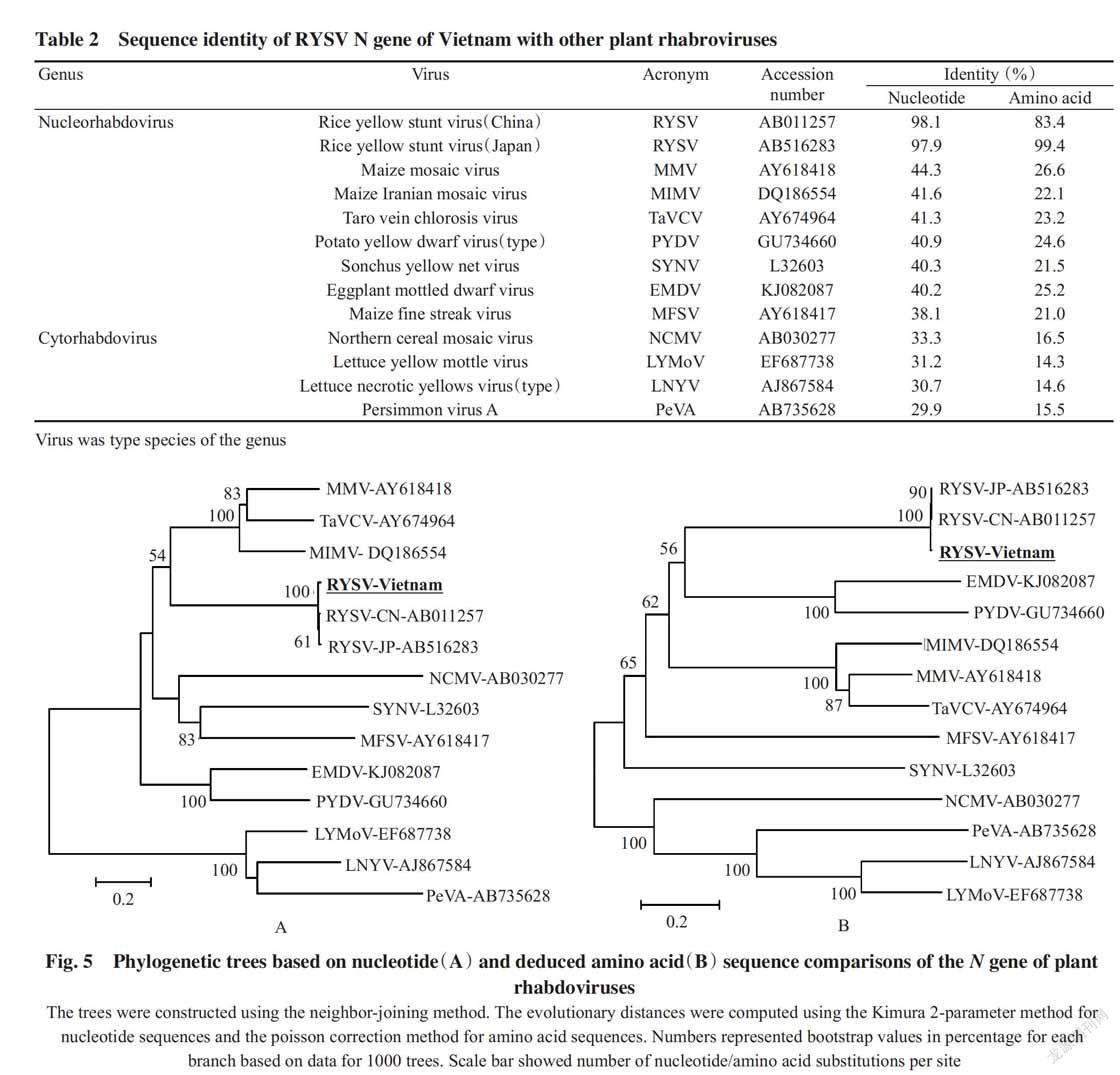
The sequence identities of the N gene of RYSV isolate from Vietnam were 98.1% and 97.9%(nucleotide level) and 83.4% and 99.4%(amino acid level) compared with those from China and Japan isolates,respectively,but much lower compared with those from other plant rhabdoviruse,ranging from 29.2% to 44.3%(nucleotide level) and from 15.5% to 26.6%(amino acid level)(Table 2). The phylogenetic ana-lysis based on the nucleotide and deduced amino acid sequences of the N gene showed three RYSV isolates from Vietnam,China and Japan formed a species cluster distinct to other plant rhabdoviruses(Fig.5),which was consistent with the sequence comparison.
2. 3 Expression of the N gene
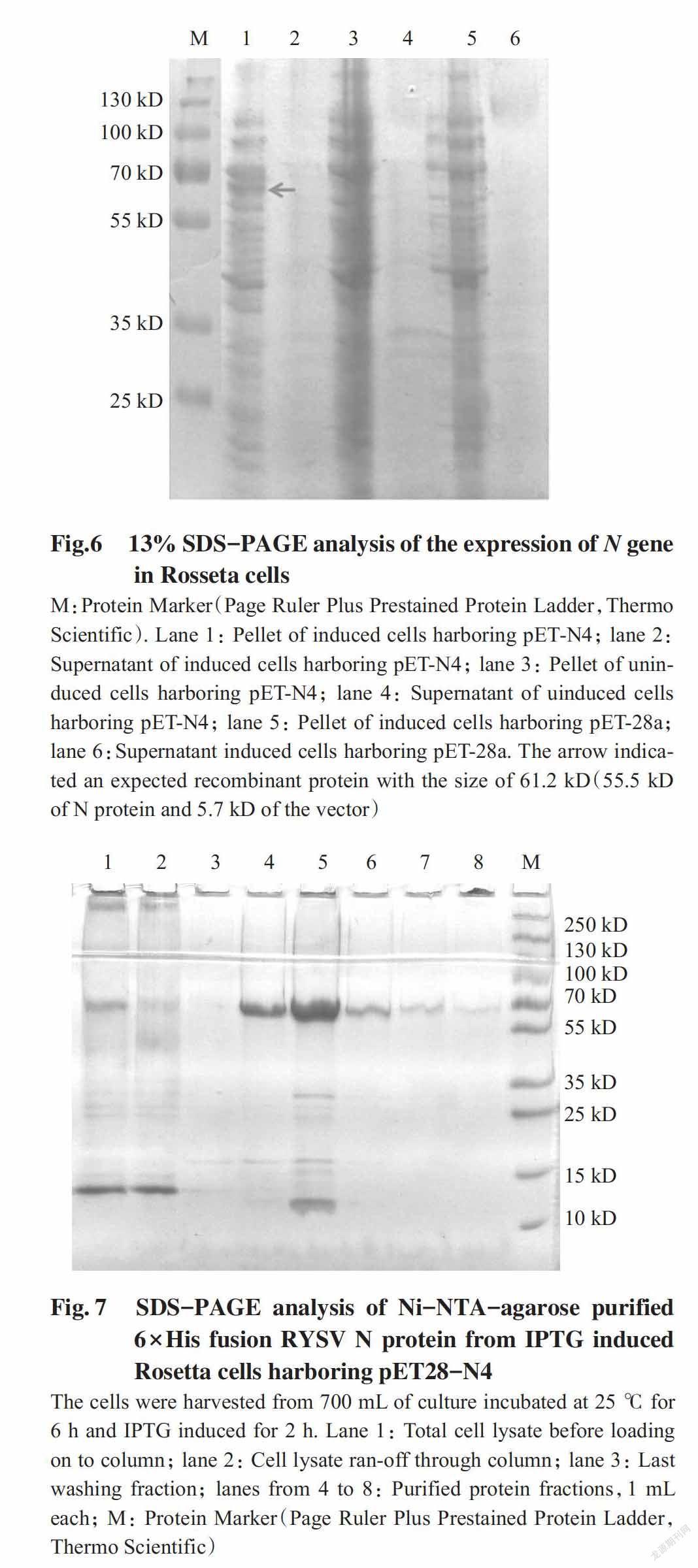
After verifying the sequence,the plasmid pET-N4 was transformed into BL21(DE3) cells of E. coli for expression. The SDS-PAGE analysis did not detect any expected products from BL21(DE3) cells induced with IPTG(data not shown). The pET-N4 plasmid was then transformed into the Rosetta(DE3) host strain. SDS-PAGE analysis detected a product with the expected size of 61.2 kD(55.5 kD of the N protein and 5.7 kD of the vector) from the IPTG induced Rosetta cells harboring pET-N4. No expected products were detected in SDS-PAGE analysis from the uninduced Rosetta cells harboring pET-N4,the induced/uninduced Rosetta cells harboring pET-28a,and the growth medium(Fig.6).
The recombinant fusion protein containing 6×His residues at N-terminal remaining in the induced Rosetta cells was purified using nickel affinity chromatography(Ni-NTA-agarose) under native conditions. The expected bands from all eluted fractions were obtained from SDS-PAGE analysis(Fig.7). Although few non-specific bands still remained in two first eluted fractions but the major purpose of the expre-ssion was to prepare the antigen of RYSV for immunization,so no further optimization of purification was done. The mixture of eluted fractions was dialyzed in PBS buffer and used as antigen. The expec-ted bands were also cut out from polyacrylamide gel,homogenized in PBS buffer and used as another antigen source.
2. 4 Production of antisera
The purified virus,the eluted fraction of the N protein and the homogenate of the polyacrylamide slices containing N protein band were used as antigens to immunize rabbits. At two and four weeks after the first injection(WAFI),0.5 mL blood was taken from the marginal ear vein of immunized rabbits and analyzed by PTA-ELISA for detecting the pre-sence of RYSV-specific antibodies in the antisera. OD405 of the antisera taken at 2 WAFI were 0.584,1.578 and 0.388 for infected tissues but only 0.181,0.281 and 0.296 for healthy tissues,respectively,indicating the presence of RYSV-specific antibodies in all three antisera. At 4 WAFI,the RYSV-specific antibodies in all three antisera increased remarkably when OD405 were as high as 1.449,2.337 and 1.649 for infected plants but only 0.375,0.294 and 0.283 for healthy plants(Table 3). All the rabbits were killed at 5 WAFI for harvesting blood. Further evalua-tion of the harvested antisera showed the antisera from rabbits immunized with eluted fraction of N protein or homogenate of the polyacrylamide slices containing N protein band were able to detect RYSV in infected plants at the dilution that was as high as 1∶10000(optimum 1∶1000); while the antiserum of the rabbit immunized with purified virus had much lower titer which was 1∶2000(optimum 1∶500)(data not shown). Because the antiserum of the rabbit immunized with purified virus had low titer and high background which indicated the presence of non-specific antibodies that reacted with plant proteins,the polyclonal antibody was then purified from the antiserum using the protein A chromatography after a step of removing the non-specific antibodies as described in the method.
2. 5 Detection of RYSV from insect vector
The antibody from the rabbit immunized with the purified virus when tested on 6 vector samples (equivalent to 6 single insects) collected in a heavily infected rice field gave the absorbance values ran-ging from 0.352 to 0.701,which were all conclu-ded as positive based on absorbance values(Table 4). All of these insect samples were also positive in RT-PCR test(Table 4 and Fig.8),indicating high specificity and sensitivity of the antibody. Similarly,the antiserum from rabbit immunized with eluted fraction of the N protein was able to detect easily the virus in single vectors with absorbance values ranging from 0.318 to 1.672(Table 4).
3 Discussion
Purification of plant rhabdoviruses was difficult because virions were enveloped and rather unstable in comparison to most other plant viruses(Jackson et al.,2018). The researchers adopted a protocol of purification of Cynodon chlorotic streak virus,a nucleorhabdovirus,from maize,a monocot plant(Lockhart et al.,1985),which was assumed to be similar to RYSV and rice with regard to purification. The presence of the enveloped viral nucleocapsis particles in the purified preparations and formation of specific antibodies in the antiserum proved this protocol was satisfactory for purifying RYSV from rice leaf tissue. However,the initial ELISA tests(data not shown) showed the produced antiserum still contained a large quantity of non-specific antibodies,suggesting that the purification process did not completely remove the plant proteins. It has been known that virus preparations purified from the infected plant materials were normally contaminated with host proteins. Therefore,the polyclonal antibodies raised from them often contained nonspecific antibodies which reacted with healthy plant extracts(Lima et al.,2012; Jackson et al.,2018).
To minimize cross reaction of antiserum to plant protein,the N protein of RYSV was selected as antigen because this protein was equivalent to coat protein that was normally used in production of polyclonal antibodies for diagnosis of plant viruses(Lima et al.,2012; Souiri et al.,2014). To verify the sequence of the cloned N gene before expression,the N gene of RYSV from Vietnam was compared with that from China and Japan. Surprisingly,while the N protein from Vietnam isolate was almost identical with that from Japan isolate,it was totally distinct in the C terminal sequence to that from China. Analysis of the sequences indicated a sequencing error which resulted in a frameshift and a termination codon change in the N gene of isolate from China. The highly sequence similarity of the N protein among three isolates after correction of the sequencing error suggested the antisera raised against this protein could detect RYSV in different countries.
Several factors affected the expression of fo-reign proteins in E. coli such as promoter strength,codon usage/bias,efficiency of ribosome binding,stabi-lity and location of the foreign protein in cells,the metabolic state of the cell(Khow and Suntrarachun,2012; Rosano and Ceccarelli,2014). The initial fai-lure of expression of the N gene cloned in pET-28a vector in the BL21(DE3) cells suggested that this E. coli strain did not support expression of the N gene of RYSV because this virus was adapted to multiply in eukaryotic cells like those of rice and insect vector. Replacement of host strain to the Rosetta(DE3) resulted in high level of expression of the N protein. The Rosetta(DE3) strain was a BL21 derivative that was designed to enhance the expression of eukaryo-tic proteins that contained codons rarely used in E. coli such as AUA,AGG,AGA,CUA,CCC,and GGA(Gopal and Kumar,2013; Rosano and Ceccarelli,2014). Thus,this codon bias-adjusted strain demonstrated to be a suitable host strain for expression of the N protein of RYSV.
In this study,antiserum produced from the homogenate of polyacrylamide slices containing N protein band was comparable to that from the eluted fraction of purified N protein regarding to the specificity and titer. An epitope,also known as antigenic determinant,of protein antigens was either linear (continuous) or conformational(discontinuous),depending on whether the amino acids that formed the epitope were contiguous in the peptide chain or not (van Regenmortel,2014). While the protein molecules in the polyacrylamide gel contain only linear epitopes,those in purified preparations contain both linear and conformational epitopes. The ELISA results,thus,indicated that most of the specific antibo-dies were induced from the linear epitopes in the N protein.
Direct immunization with slices of polyacry-lamide gel containing protein was recommended for production of antibodies(Amero et al.,2002) but it has not been applied for plant viruses. Hence,the use of the polyacrylamide slices containing N protein band offered a simplified procedure for production of antigen to produce of antibody for diagnosis of RYSV. As far as the researchers know,this method was applied for the first time for production of a plant virus. Because the detection of RYSV in the insect vector was essential to effective management of disease,the ability of the antisera and purified antibody to detect the virus in the vectors was evalua-ted. The ELISA results revealed that both antisera and purified antibody had high specificity and sensitivity that were comparable to RT-PCR in detection of RYSV.
4 Conclusion
The antisera produced by immunization of rabbits with the purified virion and the N protein of RYSV were able to detect this virus in rice plant and in single insect vector. They should be useful for epidemiological studies of this virus in Vietnam.
References:
Amero S A,James T C, Elgin S C. 2002. Production of antibodies using proteins in gel bands[M]//Waker J M. The protein protocols handbook. The 2nd Edition. Totowa:Humana Press.
Dietzgen R,Calisher C,Kurath G,Kuzmin I,Rodriguez L,Stone D. 2011. The Family Rhadoviridae[R]// King A M Q,Lefkowitz E,Adams M J,Carstens E B. Virus taxonomy: Classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses. Amsterdam: Elsevier.
Fang R X,Wang Q,Xu B Y,Pang Z,Zhu H T,Mang K,Gao D M,Qin W S,Chua N H. 1994. Structure of the nucleocapsid protein gene of rice yellow stunt rhabdovirus[J]. Virology,204(1): 367-375.
Gopal G J, Kumar A. 2013. Strategies for the production of recombinant protein in Escherichia coli[J]. The Protein Journal,32(6): 419-425.
Ha V C,Le V H,Nguyen V H,Vu T M. 2010. Identification of causative agent of rice yellow stunt disease in Bac Giang Province in summer season in 2010[J]. Journal of Plant Protection(in Vietnamese),228: 24-31.
Hibino H. 1996. Biology and epidemiology of rice viruses[J]. Annual Review of Phytopathology,34: 249-274.
Hiraguri A,Hibino H,Hayashi T,Shimizu T,Uehara-Ichiki T,Omura T, Sasaya T. 2010. Complete sequence analysis of rice transitory yellowing virus and its comparison to rice yellow stunt virus[J]. Archives of Virology,155: 243-245.
Huang Y W,Zhao H,Luo Z L, Chen X Y, Fang R X. 2003. Novel structure of the genome of rice yellow stunt virus: I-dentification of the gene 6-encoded virion protein[J]. Journal of General Virology,84: 2259-2264.
Inoue H,Nojima H,Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids[J]. Gene,96(1900): 23-28.
Jackson A O, Francki R I B,Zuidema D. 1987. Biology,structure,and replication of plant rhabdoviruses[M]//Wagner R R. The rhabdoviruses. Berlin: Springer.
Jackson A O,Dietzgen R G,Goodin M M, Bragg J N,Deng M. 2005. Biology of plant rhabdoviruses[J]. Annual Review of Phytopathology,43: 623-660.
Jackson A O,Dietzgen R G,Goodin M M,Li Z. 2018. Develop-ment of model systems for plant rhabdovirus research[J]. Advances in Virus Research,102: 23-57.
Khow O,Suntrarachun S. 2012. Strategies for production of active eukaryotic proteins in bacterial expression system[J]. Asian Pacific Journal of Tropical Biomedicine,2(2): 159-162.
Larkin M A,Blackshields G,Brown N P,Chenna R,Mc Gettigan P A,McWilliam H,Valetin F,Wallace I M,Wilm A,Lopez R,Thompson J D,Gibson T J,Higgins D G. 2007. ClustalW and ClustalX version 2.0[J]. Bioinforma-tics,23: 2947-2948.
Le D T,Netsu O,Uehara-Ichiki T,Shimizu T,Choi I R,Omura T, Sasaya T. 2010. Molecular detection of nine rice viruses by a reverse-transcription loop-mediated isothermal amplification assay[J]. Journal of Virological Methods,170(1-2): 90-93.
Lima J A A,Nascimento A K Q,Radaelli P,Purcifull D E. 2012. Serology applied to plant virology[M]//Al-Moslih M. Serological diagnosis of certain human,animal and plant diseases. London: IntechOpen.
Lockhart B E L,Khaless N,Maataoui M E L,Lastra R. 1985. Cynodon chlorotic streak virus,a previously undescribed plant rhabdovirus infecting Bermuda grass and maize in the Mediterranean area[J]. Phytopathology,75:1094-1098.
Luo Z,Chen X,Gao D. Fang R. 1998. The gene 4 of rice yellow stunt rhabdovirus encodes the matrix protein[J]. Virus Genes,16(3): 277-280.
Luo Z L,Fang R X. 1998. Structure analysis of the rice yellow stunt rhabdovirus glycoprotein gene and its mRNA[J]. Archives of Virology,143(12): 2453-2459.
Mowat W P,Dawson S. 1987. Detection and identification of plant viruses by ELISA using crude sap extracts and unfractionated antisera[J]. Journal of Virological Methods,15: 233-247.
Ou S H. 1985. Rice diseases[M]. The 2nd Edition. Surrey:Commonwealth Mycological Institute Publication.
Rosano G L,Ceccarelli E A. 2014. Recombinant protein expression in Escherichia coli: Advances and challenges[J]. Frontiers in Microbiology,5: 172.
Rybicki E,von Wechmar M,Burger J. 1990. Monospecific antibody preparation for use in the detection of viruses[M]//Burnett P A. World Perspectives on Barley Yellow Dwarf. Mexico: International Maize and Wheat Improvement Center.
Souiri A,Zemzami M,Amzazi S,Ennaji M M. 2014. Polyclonal and monoclonal antibody-based methods for detection of plant viruses[J]. European Journal of Scientific Research,123: 281-295.
Takahashi Y,Omura T,Hayashi T,Shohara K,Tsuchizaki T. 1988. Detection of rice transitory yellowing virus(RTYV) in infected rice plants and insect vectors by simplified ELISA[J]. Annals of the Phytopathological Society of Japan,54: 217-219.
Tamura K, Stecher G,Peterson D,Filipski A,Kumar S. 2013. MEGA6: Molecular evolutionary genetics analysis version 6.0[J]. Molecular Biology and Evolution,30: 2725-2729.
Uehara-Ichiki T,Shiba T,Matsukura K,Ueno T,Hirae M, Sasaya T. 2013. Detection and diagnosis of rice-infecting viruses[J]. Frontiers in Microbiology,4: 289.
van Regenmortel M H. 2014. Specificity,polyspecificity,and heterospecificity of antibody-antigen recognition[J]. Journal of Molecular Recognition,27(11): 627-639.
Wang Q,Chen X,Luo Z,Fang R. 1999. Sequence analysis of leader and trailer regions of rice yellow stunt rhabdovirus and characterization of their in vivo transcripts[J]. Scien-ce in China. Series C:Life sciences,42:50-56.
(責任编辑 陈德元)
